
Abstract
Age-related diseases pose great challenges to health care systems worldwide. During aging, endothelial senescence increases the risk for cardiovascular disease. Recently, it was described that Phosphatase 1 Nuclear Targeting Subunit (PNUTS) has a central role in cardiomyocyte aging and homeostasis. Here, we determine the role of PNUTS in endothelial cell aging. We confirm that PNUTS is repressed in senescent endothelial cells (ECs). Moreover, PNUTS silencing elicits several of the hallmarks of endothelial aging: senescence, reduced angiogenesis and loss of barrier function. Findings are validate in vivo using endothelial-specific inducible PNUTS-deficient mice (Cdh5-CreERT2;PNUTSfl/fl), termed PNUTSEC-KO. Two weeks after PNUTS deletion, PNUTSEC-KO mice present severe multiorgan failure and vascular leakage. Transcriptomic analysis of PNUTS-silenced HUVECs and lungs of PNUTSEC-KO mice reveal that the PNUTS-PP1 axis tightly regulates the expression of semaphorin 3B (SEMA3B). Indeed, silencing of SEMA3B completely restores barrier function after PNUTS loss-of-function. These results reveal a pivotal role for PNUTS in endothelial homeostasis through a SEMA3B downstream pathway that provides a potential target against the effects of aging in ECs.
Similar content being viewed by others

Introduction
Cellular aging is characterized by hallmarks such as epigenetic changes, genomic instability, mitochondrial stress and cellular senescence that promote a certain number of processes leading to physiological dysfunction and multiple pathologies1. The progressive aging of the population in Western countries has led to an increase of age-related diseases, a new health care challenge2. Of those, cardiovascular diseases (CVD) have the highest prevalence in the elderly, both in women and men. Therefore, aging is the principal risk factor for cardiovascular diseases, which is also reflected in for instance the Framingham risk score, in which the relative contribution of age is at least three times higher than all other risk factors3,4.
The vascular endothelium serves as a barrier between the blood and the surrounding tissues, and has other critical functions for homeostasis, such as maintaining vascular tone and controlling platelet activity, leukocyte adhesion and angiogenesis5. Aged endothelial cells present lower proliferative and migration capacity, higher sensitivity to apoptotic signals and cellular senescence6,7,8. As a result, endothelial aging is correlated with impaired angiogenesis, reduced capillary density, increased vascular permeability and a pro-thrombotic phenotype in vascular beds, all leading to CVD9,10.
Vascular permeability is controlled by different mechanisms involving intrinsic and extrinsic factors. Among these, there are changes in the composition of intercellular adherens and tight junctions11,12, the contractile function of the actin-myosin cytoskeleton13, paracrine signaling14 and variations in calcium transients15. An important regulator of vascular integrity is the family of axon guidance proteins, specifically the semaphorin subfamily. Initially identified as determinants of axon growth and polarization, semaphorins are shown now to have an important role in regulating vascular patterning and endothelial function16,17,18.
Phosphatase 1 Nuclear Targeting Subunit (PNUTS, PPP1R10) is a nuclear protein described to act as a binding platform for the PP1 phosphatase complex19. PNUTS is implicated in cell cycle, cancer cell proliferation and protection against DNA damage response and apoptosis20. Recently, we showed that PNUTS expression is markedly repressed in the heart during aging and CVD, and the mechanism by which PNUTS downregulation induces post-ischemic heart failure includes telomere attrition in cardiomyocytes, a hallmark of aging21. PNUTS protein contains multiple binding domains, serving as partner for several targets, as PP1, TRF2, PTEN and nucleic acids, which determine PNUTS function in different contexts22,23,24. The PNUTS-PP1 complex is formed by interaction through the canonical RxVF motif. By binding to PP1, PNUTS is implicated in the dephosphorylation and activity of several PP1 targets, such as RNA polymerase II25, Myc26, Rb27 and Spt528. miR-34a, a regulator of PNUTS expression21, has been linked to endothelial permeability and tight junctions29,30. However, whether PNUTS plays a role in endothelial aging or barrier function is not known.
Here, we show that PNUTS is an aging-regulated gene in the endothelium, and that PNUTS depletion mimics an aging endothelial phenotype. Also, we describe an endothelial-specific PNUTS knockout animal model which presents severe endothelial dysfunction similar to PNUTS depletion in cultured human endothelial cells. Particularly, endothelial PNUTS loss triggers a dramatic increase in endothelial permeability, but has no effect on adherens junction composition. RNA-sequencing shows how PNUTS loss leads to a dysregulation of gene expression, particularly in axon guidance genes. Finally, using the PP1-binding mutant PNUTSW401A, we show that aberrant SEMA3B expression is the main mediator of the loss of barrier function after PNUTS depletion in a PP1-dependent manner.
Methods
Cell culture and cellular assays
Human umbilical vein endothelial cells (HUVEC) were purchased from Lonza and cultured in endothelial cell medium containing supplements and 5% FBS (ECM; ScienceCell). Cells were cultured at 37 °C with 5% CO2. HUVECs were transfected with control or PNUTS siRNA for 48 h (except for sprouting assays, see below). For proliferation assays, cells were incubated with 10 μM EdU for 4 h using the Click-iT EdU microplate Kit (Invitrogen) according to the manufacturer’s protocol. Apoptosis was assessed by incubating the cells with 200 nM Staurosporin or medium for 4 h and the caspase 3/7 activity was assayed using the ApoOne Caspase 3/7 Assay (Promega). PP1 was inhibited by stimulation with Tautomycetin (166 nM, R&D Systems). Senescence associated β-Galactosidase activity was analyzed with the Senescence Associated β-Galactosidase Staining Kit (Cell Signaling Technologies). Images were taken with a bright-field microscope (Axio Observer Z1.0 microscope, Zeiss) and the number of total cells, as well as the number of stained cells was determined in 4 images for per condition and experiment.
Transfection and lentiviral overexpression
HUVECs were transfected with siRNAs (10nmol/l) with lipofectamine RNAiMax (Life Technologies, Carlsbad, CA), as described before31. The siRNAs used were siPNUTS (Sigma-Aldrich Hs01_00133264), siSEMA3B (Dharmacon, J-007754-09-0005) and siMYC (Dharmacon L-003282-02-0005), siRNA Universal Control 1 (Sigma-Aldrich SIC001) was used as control. Lentiviral overexpression of PNUTS was achieved by cloning the full-length human cDNA of PNUTS into pLenti4/v5-DEST (Life technologies). The PP1 non-binding mutant PNUTS (W401A) was kindly provided by Dr. Thomas Küntziger20 and cloned into pLenti4/v5. All these PNUTS sequences were modified by directed mutagenesis to introduce 3 silent mutations in the seed sequence of the siRNA against PNUTS (see Table 1 for primers). pLenti-CMV-MYC was provided by Dr. Linda Penn26. Lentiviral particles were generated as described earlier32.
Sprouting angiogenesis and immunofluorescence
Endothelial angiogenesis was studied by spheroid sprouting assay. HUVECs were first transfected with the indicated siRNAs for 24 h. Then, cells were trypsinized and added to a mixture of culture medium and methylcellulose (80%:20%) and transferred to a 96-well plate to allow the formation of spheroids for 24 h at 37 °C. The spheroids were collected, added to methylcellulose with FBS (80%:20%) and embedded in a collagen type I (BD Biosciences) gel. Following incubation for 24 h at 37 °C with 50 ng/ml VEGF the gels were fixed with formaldehyde and microscope images were taken at 10x magnification (Axio Observer Z1.0 microscope, Zeiss). The cumulative length of sprouts was quantified using image analysis software (AxioVision 4.8, Zeiss). For immunofluorescence (IF), 48 h after transfection cells were fixed with PFA 4% and incubated with anti-CD31 (1:40, BD Biosciences 550389) or anti-VE-Cadherin (1:400, CST 2500XP), and actin fibers were stained with Acti-stain 555 (Cytoskeleton Inc). Images were obtained in a Nikon A1R confocal microscope. Endothelial gaps were measured by quantifying the intercellular area versus the total area in 4 fields per image, 3 images per experiment.
Endothelial cell impedance and transwell assays
After lentiviral transduction and/or transfection, HUVECs were plated in 96-well arrays (96W10idf, Applied Biophysics) at 40,000 cells/well and endothelial cell impedance was measured using the multi-frequency mode for 48 h (ECIS Zθ, Applied Biophysics). Endothelial multifrequency resistance was modeled for the calculation of cell-cell adhesion (Rb). Transwell assay was performed as explained elsewhere33.
Flow cytometry
Cell surface expression of VE-Cadherin and PECAM1 was analyzed in HUVECs by flow cytometry. Briefly, transfected HUVECs were detached with Accutase and washed in cold incubation buffer (0.1% BSA in PBS). Cells were blocked (5% BSA in PBS) for 30 min on ice and subsequently incubated with fluorophore-labeled antibodies for 30 min on ice. Protein cell surface expression was analyzed using a FACSCalibur™ device (BD Biosciences).
qRT-PCR and RNA Sequencing
Total RNA from cultured cells was isolated with Direct-zol RNA miniprep (Zymogen) and from tissues using miRNeasy Mini kit (Qiagen) according to the manufacturer’s protocols. Intima RNA was isolated by flushing 200 μl of Trizol through the lumen of isolated aortas. Total RNA was reverse transcribed using random hexamer primers and iScript cDNA synthesis kit (BioRad). qPCR was performed with SYBR Green Master Mix on CFX384 Real Time PCR Detection System (BioRad), using primers listed in Table 1. Gene expression analysis was done using the 2-ΔCT method. RNA obtained from HUVECs and mouse lung tissue was sequenced by Exiqon (whole transcriptome sequencing, 50 bp, 30 M reads) and data analysis was performed using DAVID functional annotation tools and Gene Set Enrichment Analysis.
Western blot
Total cell lysates were collected 48 h after HUVEC transfection or 5 days after lentiviral treatment and western blot (WB) carried as described before34. The primary antibodies used were anti-PNUTS (R&D Systems, AF21581), anti-CD31 (Invitrogen, 37-0700), anti-VE-Cadherin (Sigma-Aldrich, V1514), anti-MYC (CST, 9402), anti-phospho-MYC-T58 (abcam, ab185655), anti-tubulin (Thermo Fisher, RB-9281-P) and anti-β-actin (Sigma-Aldrich, A-5441). Uncropped Western blots are depicted in Supplementary Fig. S7.
Polysome profiling
siControl and siPNUTS-transfected cells were trypsinized, pelleted and snap-frozen. Polysome profiling was performed as previously described35. Briefly, cell pellets were lysed in polysome lysis buffer (gradient buffer containing 100 mM KCl, 10 mM MgCl2, 0.1% NP-40, 2 mM DTT, and 40 U/ml RNasin; Promega, Leiden, Netherlands), and onto 17–50% sucrose gradients and ultracentrifuged for 2 h at 40,000 rpm. The gradients were displaced into a UA6 absorbance reader and absorbance was recorded at an OD of 254 nm.
ELISA
HUVECs were transfected with siRNA in a 6 well plate as described above. The transfection mix was replaced with 1 ml full medium 4 h post transfection. Supernatants were collected 72 h post transfection and spun down (1000 × g, 20 min at 4 °C) to eliminate cell debris. In order to quantify SEMA3B levels, 300 µl supernatant was applied to a human SEMA3B ELISA plate (DLdevelop, Shuigoutou, China), the SEMA3B protein standard was diluted in full medium. The following procedure was performed according to the manufacturer’s instructions.
Animal procedures
All mice experiments were carried out in accordance with the principles of laboratory animal care as well as according to the German and Dutch national laws. The studies have been approved by the local ethical committees and performed in accordance with the ethical standards laid down in the Declaration of Helsinki. PNUTSflox/flox mice were generated by inserting LoxP sequences, flanking Ppp1r10 exons 9 to 14, at 69 bp downstream of exon 8 and 139 bp upstream exon 15 (Genoway, France). PNUTSflox/flox mice were crossed with the Cdh5-CreERT2 line to obtain the endothelial-specific inducible PNUTS knockout strain36. Genotyping was performed using primers from Table 1. Cdh5-CreERT2xPnutsflox/flox (PNUTSEC-KO) and Pnutsflox/flox (WT) littermates were injected intraperitoneally with 2 mg/day tamoxifen dissolved in peanut oil (Sigma-Aldrich) for 5 consecutive days and on the 8th and 10th day after the first injection, and then sacrificed for organ harvest at day 15th. All mice were 2–12 months old at the start of the experiments and the total number of mice used in this study is 15 mice per group. Aortic rings were obtained as described37 and incubated for 48 h with 10 ng/μl VEGF. After fixation, images were taken at 5x magnification and stitched with a Zeiss Axiovert 100 and the endothelial sprouts length were quantified with an image analysis software (AxioVision 4.8, Zeiss). The Evans Blue Extravasation assay was performed on day 14th adapting the protocol by Radu et al. 38 Briefly, PNUTSEC-KO and WT mice were injected in the tail vein with 25 mg/Kg of Evans Blue solution and observed for 1 h before euthanasia. Then, organs were harvested and incubated for 48 h in formamide to extract the extravasated dye, which was quantified in a spectrophotometer against a standard curve of known concentrations of Evans Blue.
Histology
Organs were fixed in formalin and embedded in paraffin. 4 μm sections were obtained and stained with Periodic-Acid Schiff (PAS, Sigma-Aldrich, 395B) and hematoxylin-eosin. Images were obtained in a Leica microscope. Kidney sections were antigen retrieved with citrate buffer and immunostained with an anti-CD31 antibody (1:20, Dianova, DIA-310). Images were taken in a Nikon A1R confocal microscope. Assessment of the capillary, mesangial and glomerular areas was performed on PAS-staining kidney sections using ImageJ software. The capillary surface area was defined as the surface area within the glomerulus tuft covered by capillary loops. The mesangial surface area was defined as a PAS–positive and nuclei-free area in the mesangium. Both quantifications were based on 8 glomeruli per group.
Statistics
GraphPad 7 (GraphPad Software) was used for statistical analyses. Comparison of two different conditions was analyzed by two-tailed Student’s t test or Mann–Whitney, multiple comparisons were performed by one-way ANOVA using Dunnett’s, Bonferroni or Tukey’s correction. Data are expressed as means ± SEM, p < 0.05 was considered as statistically significant (*p < 0.05; **p < 0.01; ***p < 0.001). The sample size n states the number of independent experiments, unless denoted otherwise. All results were reproduced in at least three technically independent replicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
PNUTS KD induces partial senescence of endothelial cells in vitro
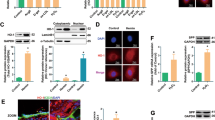
The first question we sought to answer was whether PNUTS is repressed during aging in the endothelium, as we had described previously for the myocardium, where PNUTS regulates telomere length through TRF2, one of its binding partners21. Therefore, we used a model of serial passaging of HUVECs39,40. PNUTS expression was repressed by 50% in this cell culture-induced aging model (Fig. 1a). Furthermore, we assessed Pnuts mRNA expression in the aortic intima of young (8—12 weeks old) and aged mice (18–20 months old), showing a marked reduction of Pnuts expression in the aortic endothelium (Fig. 1b). Consequently, we hypothesized that PNUTS may have a role in maintaining endothelial homeostasis which is lost during aging. For this purpose, we used an siRNA-based knockdown (KD) strategy in HUVECs. We selected an siRNA against PNUTS which caused a decrease of PNUTS protein levels of nearly 90% (Fig. 1c). Loss of PNUTS provoked an increase in the expression of p21, p16INK4A, β-galactosidase activity and several senescence-associated secretory protein genes (SASP), but not all SASP genes we measured (Fig. 1d, e, S1b). Next to senescence, aging can be accompanied by an increase in apoptosis and a decrease in proliferation. Accordingly, PNUTS KD significantly decreased proliferation (Fig. 1f), while apoptosis was not affected (Fig S1a). However, PNUTS KD increased cell susceptibility to a pro-apoptotic stimulus (Fig S1a), one of the hallmarks of endothelial aging8. These results indicate a role for PNUTS in partial inhibition of endothelial cell senescence and aging.

a HUVECs were cultured for 3 or 17 passages (P) and PNUTS expression was measured by qRT-PCR (n = 4). b Aortic intima was isolated from young (8–12 weeks old) and aged (18–20 months old) mice and immediately lysed for RNA isolation. Intima RNA was sequenced. c–f HUVECs were transfected with siRNAs targeting PNUTS or a control sequence. c PNUTS protein levels were determined by WB at 48 h after transfection, relative to alpha-tubulin. Densitometric quantification is depicted on the right (n = 7). d Changes in p21 expression were assessed by qRT-PCR. Expression values are relative to siControl-treated HUVECs and normalized to RPSA mRNA (n = 6). e The percentage of senescent HUVECs was analyzed by staining for senescence-associated β-Galactosidase (SA-(β-Gal) 72 h after transfection. Images were taken (4 fields per sample) and the percentage of senescent cells (blue) over the total number of cells in each field was calculated in n = 3 independent experiments, 2–3 biological replicates per group and experiment). f Cell proliferation was assayed by testing the incorporation of EdU after transfection (n = 4). *p < 0.05, **p < 0.01, ***p < 0.001. Error bars depict the standard error of the mean (SEM).
PNUTS is necessary for endothelial cell sprouting in vitro and in vivo
As angiogenesis is typically reduced during aging10, we performed a spheroid sprouting assay to check the angiogenic capability of PNUTS KD HUVECs. The results indicate that the total length of sprouts, from the surface of the spheroid to the tip cell, was similar in control and KD cells, but in the latter the sprouts showed an aberrant pattern characterized by numerous discontinuities (Fig. 2a, b). These results suggest that loss of PNUTS induces loss of stalk cell function or cell-cell adhesions.

a In vitro sprouting was analyzed under VEGF (50 ng/ml) stimulation. Representative images are shown, red arrows indicate discontinuous sprouts. b Quantification of cumulative sprout length (left) and number of discontinuous sprouts (right) (n = 30 spheroids per group in 3 independent experiments were measured). c Endothelial Pnuts expression of PNUTSEC-KO and WT mice was assessed by RT-qPCR in intima samples 15 days after initiating tamoxifen treatment and normalized to Rplp0 mRNA (n = 3). d In vivo angiogenesis was tested by aortic ring assay in WT and PNUTSEC-KO mice in basal conditions or upon 48 h of VEGF (10 ng/ml) stimulation. e Quantification of aortic ring sprouting. n = 4–5 mice per group. *p < 0.05, **p < 0.01, ***p < 0.001. Error bars depict the standard error of the mean (SEM).
To assess the physiological role of PNUTS in ECs we sought to use an in vivo model of PNUTS loss. To this moment, no loss-of-function mouse model had been described for PNUTS, therefore we generated an endothelial-specific inducible PNUTS knockout mouse line, Cdh5-CreERT2xPnutsflox/flox (PNUTSEC-KO), which undergoes efficient loss of PNUTS in the endothelial layer two weeks after tamoxifen treatment compared to PNUTSfl/fl (WT) (Fig. 2c, Fig S2a, b). Using this mouse model, we assessed the angiogenic capability of ECs lacking PNUTS. PNUTSEC-KO aortas were subjected to an aortic ring assay, which showed that the sprouting of aortic rings in PNUTSEC-KO mice was abrogated compared to WT (Fig. 2d, e), confirming the findings obtained in vitro and suggesting a role for PNUTS in maintaining endothelial cell function.
PNUTS EC-knockout mice present multiorgan failure and vascular leakage
We carefully monitored PNUTSEC-KO mice after initiation of PNUTS excision by tamoxifen treatment to assess the physiological role of endothelial PNUTS. Within 15 days after tamoxifen treatment, all PNUTSEC-KO mice died, accompanied by a severe accumulation of fluids in peritoneal and pleural cavities. Pathological analysis of kidneys, lungs and hearts showed that PNUTSEC-KO mice suffered from glomerulosclerosis, capillary dilation and pulmonary and cardiac edema (Fig. 3a). Image analysis of PAS-stained kidney sections showed that PNUTSEC-KO mice present a marked decrease in the number and area of capillary loops as well as an expansion of the mesangial area and fraction in their renal glomeruli (Fig. 3a). As our findings pointed that the loss of PNUTS induced a major change in the phenotypical characteristics of ECs, we assessed global gene expression by RNA sequencing (RNA-seq) in lungs of PNUTSEC-KO and WT mice (Fig. 3b, Supplementary data 2). The RNA-seq data showed an increase in inflammatory response and coagulation pathways and changes in genes related to endothelial cell-cell and cell-matrix interaction. Intriguingly, while the endothelial cell markers Pecam1 and VE-Cadherin were unaltered, angiogenesis- and coagulation-related genes Vegfr2 and Thbd showed a slight but significant decreased expression, indicating a potential decrease in endothelial cell function rather than a loss of endothelial cells (Fig. 3c). p21 and several SASP genes were upregulated, suggesting a role for PNUTS in endothelial senescence also in vivo (Fig S2c). Presence of ECs in PNUTSEC-KO kidneys was confirmed by Immunofluorescence of PECAM1 in renal glomeruli and confirmed the loss of capillary organization in glomeruli, as observed in Fig. 3a. (Fig. 3d). Intriguingly, the phenotype observed in several organs, including the presence of fluid in the cavities, pointed to increased microvascular permeability. Therefore, we investigated whether PNUTSEC-KO mice suffered from vascular leakage. We performed Evans Blue extravasation assays and found that the tracer was able to cross the endothelial barrier to the tissue in several organs in PNUTSEC-KO mice, as well as the peritoneal cavity, pointing to a loss of barrier integrity in vivo (Fig. 3e, f). These results suggest that endothelial PNUTS is critical for maintenance of endothelial barrier and its loss is incompatible with life due to severe vascular leakage.

a Histopathological study of different tissues in PNUTSEC-KO mice compared to WT mice: left, heart samples (haematoxylin-eosin) presenting edema; middle, lung samples (haematoxylin-eosin) presenting thrombi; right, kidney samples (PAS staining) showing glomerulosclerosis (asterisk) and capillary dilatation (black arrow) in renal glomeruli. Quantifications of the kidney sections show the absolute and relative contribution of capillaries and mesangial tissue in glomeruli (n = 8 per group). b RNA-seq was performed with lung tissue of WT and PNUTSEC-KO mice and was analysed for differentially regulated pathways using Gene Set Enrichment Analysis. Enrichment scores of the indicated pathways are plotted on the x axis (n = 3). c The expression levels of the endothelial markers Ve-cadherin, Pecam1, Vegfr2, Thbd and Pdgfra in WT and PNUTSEC-KO lung samples were confirmed by RT-qPCR and normalized to Rplp0 mRNA (n = 3). d The presence of ECs in the glomerular capillary network was investigated by PECAM1 immunostaining in kidneys of WT and PNUTSEC-KO mice. Red and white asterisks indicate potential mesangial proliferation and glomerulosclerotic areas, respectively. e, f An Evans Blue (EB) extravasation assay was performed to measure the vascular extravasation in different organs. e Representative lung and kidney images 1 h after intravenous administration of EB at 25 mg/kg. f Colorimetric measurement of extravasated EB into lung, kidney and peritoneal fluid (n = 4–5 mice per group). *p < 0.05, **p < 0.01, ***p < 0.001. Error bars depict the standard error of the mean (SEM).
PNUTS loss compromises the endothelial barrier by disrupting cell-cell interaction
The endothelial monolayer is responsible for regulating the trafficking of solutes and cells from the blood to the surrounding tissues, forming a selective barrier between both. In light of our in vivo results, we confirmed whether PNUTS is necessary for this function. We measured the impedance of HUVEC monolayers using an electrical cell impedance system (ECIS) and found that loss of PNUTS decreased the resistance of the endothelial monolayer (Fig. 4a, b), suggesting an increase in endothelial permeability. As a confirmation of these results, we performed a transwell assay, which showed an increased leakage through the monolayer of PNUTS-silenced cells, as measured by passage of HRP (Fig. 4c). This decrease in endothelial barrier function is mainly due to a loss of endothelial cell-cell interaction, as indicated by multifrequency modeling of Rb (Fig. 4d, e). Western blot and flow cytometry measurements of HUVECs showed that the effects of PNUTS depletion on endothelial barrier function are not due to decreased levels or delocalization of adherens junction proteins (Fig. 4f, g). However, IF analysis of VE-Cadherin and PECAM1 in PNUTS-silenced HUVECs reveal the presence of junctional defects and an increase of intercellular gaps that might explain the inability to form an efficient barrier in the absence of PNUTS (Fig. 4h, S3). Altogether, this suggests that PNUTS is necessary to maintain a functional endothelial monolayer through a mechanism independent of adherens junction protein expression.

a–h HUVECs were treated with siRNA (si) targeting PNUTS or a control sequence. a-b) HUVEC barrier resistance was assessed by ECIS at 4000 Hz for 48 h, presented as average resistance ± SEM of n = 3 experiments, 8 biological replicates per group and experiment. c HUVECs were seeded in transwells and HRP passage through the endothelial monolayer was assessed by absorption measurements (450 nm) and shown as percentage of total HRP (n = 5). d, e Cell-cell interaction was assessed by modeling of data obtained by ECIS measured as Rb (Ω*cm2); presented as average resistance ± SEM of n = 3 experiments, 8 biological replicates per group and experiment. f Cell surface presence of PECAM1 and VE-cadherin was assessed by flow cytometry 48 h after siRNA transfection (n = 3). g Total PECAM1 and VE-cadherin levels were assessed by Western Blotting (WB). Tubulin and β-actin expression was used as loading control. h Cells were grown into confluence and immuno-stained for VE-cadherin, PECAM1 and F-actin. DAPI was used to stain nuclei. The presence of intercellular gaps in endothelial monolayers was measured by quantifying the intercellular areas versus the total area in 4 fields per image, 3 images per experiment, n = 4. i Schematic representation of the PNUTS lentiviral vectors used for the barrier rescue experiments. Both vectors included silent mutations in the seed sequence of siPNUTS. HUVECs were transduced with the indicated constructs for 8–10 days and later transfected with siControl or siPNUTS to silence endogenous expression of PNUTS, before subjecting them to ECIS and cell counting (4–6 independent experiments, 4 biological replicates per group and experiment). j PNUTS expression in total cell lysates of HUVECs treated with indicated vectors and siRNAs was analyzed by WB, Tubulin was used as a loading control. Representative WB is shown. k Change in cell-cell interaction measurement from ECIS 48 h after cell seeding, measured as variation of Rb of siPNUTS- vs siControl-treated cells. l Cell proliferation of ECIS-assayed HUVECs, measured as % number of cells relative to control. *p < 0.05, **p < 0.01, ***p < 0.001. Error bars depict the standard error of the mean (SEM).
Previous studies describe PNUTS as a binding partner and activator for PP122. We therefore aimed to assess the potential role of PP1 for PNUTS function by using several lentiviral vectors to overexpress siRNA-resistant wild-type (PNUTSWT) and PP1-non-binding mutant PNUTS (PNUTSW401a) (Fig. 4i). All of them proved to be resistant to siPNUTS treatment (Fig. 4j) and were used to test the relevance of the PP1-binding domain for the role of PNUTS in barrier maintenance by ECIS. As seen in Fig. 4k, only the overexpression of PNUTSWT was able to rescue the drop in barrier function caused by PNUTS KD, while the PP1-binding mutant was not. Moreover, the PP1-binding is important for endothelial survival, as the overexpression of the PNUTSW401A mutant decreased EC proliferation to the same levels as siPNUTS treatment, while PNUTSWT had a positive effect on proliferation of ECs (Fig. 4l).
PNUTS loss triggers a complex transcriptional profile change in endothelial cells
In order to more elaborately study the mechanism by which PNUTS controls EC function, we studied the gene profile elicited by PNUTS loss. We performed RNA-seq of siControl and siPNUTS-treated HUVECs. We found 7034 transcripts significantly up- or downregulated (Supplementary data 3). KEGG pathway functional annotation identified several affected pathways, among which mRNAs encoding ribosomal proteins stood out (Fig S4a). Sixty-eight ribosomal protein-encoding genes were significantly altered, of which 62 were strongly downregulated (Fig S4b). In line with this finding, a polysome profiling of siControl- and siPNUTS-treated HUVECs showed that PNUTS KD provokes a reduction of ribosomal content in ECs (Fig S4c).
Gene set enrichment analysis showed a decrease in expression of Myc-dependent targets (Fig S5). MYC has been described to have roles in endothelial proliferation and angiogenesis41 and it has been recently suggested that MYC phosphorylation and subsequent degradation might be controlled by the PNUTS26. MYC protein levels were decreased in PNUTS KD cells, while phosphorylation of MYC at Threonine 58 was increased, which induces ubiquitination and proteasomal degradation of MYC (Fig S5c). However, siRNA-mediated KD of MYC had no effect on endothelial barrier function, while lentiviral-mediated overexpression of MYC had a negative effect, opposite to expected (Fig S5d). Therefore, MYC is a negative regulator of the endothelial barrier in a PNUTS-independent manner.
PNUTS regulates barrier function via suppression of SEMA3B expression
The complexity of the data obtained by RNA-seq led us to compare the gene expression profile after PNUTS loss in HUVECs and mouse lungs. A total of 277 common genes were found to be regulated in both datasets. KEGG pathway analysis of these revealed a significant presence of axon guidance pathway genes (Fig. 5a). We confirmed the RNA-seq data by qPCR in PNUTS-silenced HUVECs, finding a dramatic induction of SEMA3B expression (Fig. 5b), one of the highest upregulated genes after PNUTS KD (Fig. 5c, Supplementary data 3). SEMA3B protein was also found to be increased in the supernatant of siPNUTS-treated cells (Fig. 5d), as quantified by ELISA. Furthermore, the expression of Sema3b in the aortic intima of PNUTSEC-KO mice is increased 7-fold compared to WT mice (Fig. 5e). SEMA3B has been described previously as a secreted protein that causes repulsion signals in ECs which interferes with correct angiogenesis42, but has not yet been linked to endothelial barrier formation.

a The two subsets of RNAseq data we obtained, from endothelial-depleted PNUTS mouse lungs and PNUTS knockdown (KD) HUVECs, were compared to find common targets of PNUTS depletion. The Venn diagram depicts the finding of 277 common transcripts, which were functionally analysed using KEGG pathways analysis, shown in the table below. b Changes in axon guidance gene expression after PNUTS silencing was assessed by RT-qPCR. Expression values are relative siControl-treated HUVECs and normalized to RPSA mRNA (n = 6). c Volcano plot showing the distribution of gene expression in PNUTS KD versus control HUVECs. SEMA3B is marked in red. d Supernatant SEMA3B concentration was determined by ELISA 72 h after PNUTS silencing (n = 4). e Expression levels of Sema3b mRNA in intima samples of PNUTSEC-KO mice was assessed by RT-qPCR, relative to WT samples and normalized to Rplp0 mRNA. f HUVECs were treated with vehicle or Tautomycetin (166 nM) for 48 h and mRNA was analyzed by RT-qPCR for expression of SEMA3B, normalized to RPSA mRNA (n = 3). g Expression of SEMA3B was measured in mRNA samples of HUVECs assayed in Fig. 4j–l, relative to control cells and normalized to RPSA mRNA. h-i HUVECs were co-transfected with siPNUTS and/or siSEMA3B were subjected to ECIS for 48 h (n = 4 independent experiments, 4 biological replicates per group and experiment). h Cell-cell interaction was modeled. i Endothelial resistance was measured at 4000 Hz. j Top panel: siSEMA3B rescues the effect of PNUTS silencing on adherence junctions (shown as PECAM1 IF staining). Bottom panel: PECAM1 IF staining shows time course of change in adherens junctions upon stimulation with recombinant human SEMA3B. k Cell-cell interaction was modeled using ECIS. The arrow indicates the 48 h time point at which recombinant SEMA3B was added to the medium (or not; untreated). Quantification was performed at 60 h (n = 4 biological replicates and 3–4 technical replicates). l Graphic summary of the proposed mechanism. In young individuals, PNUTS interacts and promotes activity of PP1, which represses the expression of SEMA3B. Endothelial cells are in homeostasis and maintain their barrier function. During aging, PNUTS is repressed in endothelial cells. The absence of PNUTS inhibits PP1 function at the SEMA3B promoter activating SEMA3B expression. SEMA3B exerts repulsive signals between endothelial cells, promoting intercellular gaps and disrupting the barrier. This provokes a series of critical changes in the cells leading to cellular senescence. *p < 0.05, **p < 0.01, ***p < 0.001. Error bars depict the standard error of the mean (SEM).
PP1 has been described to exert a broad role in regulating gene expression, thus we studied SEMA3B expression after treating HUVECs with the PP1 inhibitor drug Tautomycetin a highly specific inhibitor of PP1. Tautomycetin blocks PP1 enzymatic activity by binding PP1 Cys127, in a higher selective manner compared to other PP inhibitors as okadaic acid or TF-23A43,44. Indeed, PP1 inhibition triggered a significant induction of SEMA3B expression (Fig. 5f). Furthermore, the induction of SEMA3B expression after PNUTS silencing is rescued by overexpression of PNUTSWT, but not by overexpression of the PP1-binding mutant PNUTSW401A (Fig. 5g). Finally, we hypothesized that SEMA3B might be the causal mechanism triggered by loss of PNUTS leading to a disruption of barrier function. To test this, we co-treated HUVECs with siPNUTS and siSEMA3B and found that SEMA3B knockdown completely abolished the effect of PNUTS loss on cell-cell interaction and endothelial resistance (Fig. 5h-i) and rescued the effect of siPNUTS on adherens junctions (Fig. 5j, top panel). To further test the effect of this semaphorin on cell-cell interactions, we cultured HUVECs with medium containing recombinant human SEMA3B and found that SEMA3B induces changes in PECAM1 membrane pattern similar to those of siPNUTS (Fig. 5j, bottom panel) and that SEMA3B reduces cell-cell interactions, as measured by ECIS (Fig. 5k). Altogether, these results point to the axon repulsion signal SEMA3B as a disruptor of endothelial monolayer integrity which is repressed by PNUTS in homeostatic situations through PP1 binding and activity (Fig. 5l).
Of note, the previously described regulator of PNUTS, miR-34a, has been shown to promote endothelial permeability through targeting tight junction-related protein kinase Cε29,30. Since miR-34a plays a role in cardiac aging and disease by post-transcriptional regulation of PNUTS21, we hypothesized that miR-34a could be upstream of PNUTS and its role in endothelial permeability as well. Transfection of miR-34a in HUVECs provoked a decrease in barrier function measured by ECIS. However, overexpressing PNUTS (using a construct that does not contain a miR-34a binding site) in these cells was not able to rescue the barrier (Fig S6a). Moreover, overexpression of miR-34a only causes a mild downregulation of PNUTS in HUVECs, and not a significant increase of SEMA3B expression (Fig S6b). These results suggest that the role of PNUTS on endothelial cell aging and function is independent of miR-34a.
Discussion
This study describes the aging-regulated protein PNUTS as a critical element to maintain endothelial homeostasis, as loss of endothelial PNUTS elicits a phenotype of endothelial leakage compromising homeostasis. PNUTS, through its PP1-binding domain, is necessary for endothelial barrier integrity. Loss of PNUTS deregulates the signature gene expression profile in ECs and triggers the induction of SEMA3B, a member of the axon guidance family of proteins that acts as a repulsion signal for ECs.
PNUTS structure and function have been mainly studied in vitro in cell lines, while the role of PNUTS in physiology and disease has been largely unknown. In particular, the role of PNUTS in the cardiovascular system or aging has been described previously only in the myocardium, where PNUTS regulates telomere length through TRF2 in cardiomyocytes and is downregulated during aging or after myocardial infarction21. In the present work, we sought to assess the role of PNUTS in the endothelium. As seen in cardiomyocytes, endothelial PNUTS is aging-regulated and critical to maintain cell functionality in ECs. In fact, PNUTS loss recapitulates many of the hallmarks of endothelial aging, such as expression of senescence markers, decreased proliferation, gene expression deregulation and endothelial dysfunction (Figs. 1, 2, 4 and S1). However, PNUTS loss in the myocardium provoked an increase in DNA damage and apoptosis through the aforementioned mechanism, whilst in the endothelium PNUTS has a role in maintaining endothelial integrity via regulation of SEMA3B expression. This suggests that PNUTS function and mechanism of action is cell type- and binding partner-specific. PNUTS was initially described as a binding platform for PP1, and although several other binding partners of PNUTS have been described over time45, the main body of knowledge on PNUTS function continues to be related to this phosphatase. The PNUTS-PP1 axis has been suggested to be responsible for DNA damage response20, MYC activation26, cell cycle entry46, apoptosis27, gene expression regulation25 and transcription28. None of these studies report, however, a role for PNUTS-PP1 in physiological processes, with the exception of the work of Ciurciu and colleagues25, which modeled their findings in the embryonic development of D. melanogaster. Our work shows the contribution of PNUTS in a physiological context in mammals.
However, vascular permeability is controlled by complex mechanisms, for instance by haemodynamic changes and lymphatic drainage. These mechanisms are difficult to separate in vivo and cannot be well addressed in an in vitro setting. This is an important limitation of our study. Another limitation is that the assays we use to study endothelial permeability do not distinguish between transcellular and paracellular leakage. Even though the data suggest that SEMA3B-mediated repulsion results in paracellular leakage after reduction of PNUTS, a contribution of transcellular leakage cannot be excluded.
The decline of PNUTS protein levels with ageing has been shown before in the heart21, where miR-34a induction results in reduction of PNUTS levels. Our experiments show that miR-34a can also target PNUTS in ECs, but that this reduction is relatively mild (Fig S6). It is therefore likely that other mechanisms contribute to the decline of PNUTS levels with ageing. One potential mechanism could be the recently identified alternative splicing regulation of PNUTS that results in a non-coding transcript47, in which an increase of the non-coding RNA would inherently results in a decrease of the PNUTS mRNA. One of the functions of ECs is to form a barrier between the blood and the underlying tissues that allows correct exchange of nutrients and waste products. This important function is also impaired by aging, which induces hyperpermeability in the peripheral tissues and eventually edema formation due to an increase in transcellular and paracellular permeability48. Notably, our results in PNUTSEC-KO mice and PNUTS KD HUVECs point to endothelial barrier dysfunction independent of adherens junctions (Figs. 3a, e, f and 4a–g), which we concluded was the main consequence of PNUTS loss in the endothelium. Interestingly, even though PNUTS silencing induces gaps in between cells, these events are rather rare, and the increase in permeability seems to be restricted to smaller molecules, suggesting that dynamic repulsive signals may cause the barrier disruption. These effects are mediated by via the PP1 binding domain of PNUTS (Fig. 4h–l). Previously, PP1 had been described to have a role in endothelial permeability by dephosphorylation of cytoskeleton proteins49, but here we provide evidence that PP1 function in ECs is linked to a nuclear co-factor (PNUTS).
Hence, we considered a nuclear mechanism that provoked cells to be less prone to form proper interactions. PNUTS loss elicits a complex response in gene expression both in vivo (Fig. 3b) and in vitro (Fig S4a, S5a), which led to the identification of SEMA3B as potential target of the PNUTS-PP1 axis. This secreted semaphorin, as others of the axon guidance family of genes, was historically studied for its repulsion role in the guidance of neuronal growth in the developing neural system50, but new roles in endothelial barrier maintenance, preeclampsia and inhibition of angiogenesis have emerged17,42,51,52, which aligns with our findings. Little is known about the regulation of SEMA3B expression. Our study suggests that SEMA3B is induced in the absence of a working PNUTS-PP1 complex (Fig. 5b–g), and that it is the main actor in impeding the formation of a healthy endothelial barrier (Fig. 5h–j) upon loss of PNUTS. Finally, our results highlight a concept that explains loss of barrier function during aging in which dropping PNUTS levels result in aberrant activation of SEMA3B expression that induces repulsion of ECs.
In conclusion, this study provides evidence that PNUTS is critical for endothelial function during aging. Our data highlight the importance of PNUTS in providing a tight control of the endothelial gene expression profile, which is necessary to maintain barrier functionality through the regulation of the semaphorin family member SEMA3B, and as a potential clinical target against aging-related vascular hyperpermeability.
Data availability
The RNA sequencing data can be found at the gene expression omnibus of NCBI under accession numbers GSE263531 and GSE263742. The source data behind the graphs in the paper can be found in Supplementary data 1. All additional data are available from the corresponding author on reasonable request.
References
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Christensen, K., Doblhammer, G., Rau, R. & Vaupel, J. W. Ageing populations: The challenges ahead. Lancet 374, 1196–1208 (2009).
North, B. J. & Sinclair, D. A. The intersection between aging and cardiovascular disease. Circ. Res. 110, 1097–1108 (2012).
D’Agostino, R. B. Sr. et al. General cardiovascular risk profile for use in primary care: The Framingham Heart Study. Circulation 117, 743–753 (2008).
Vita, J. A. Endothelial function. Circulation 124, e906–e912 (2011).
Lahteenvuo, J. & Rosenzweig, A. Effects of aging on angiogenesis. Circ. Res. 110, 1252–1264 (2012).
He, T., Joyner, M. J. & Katusic, Z. S. Aging decreases expression and activity of glutathione peroxidase-1 in human endothelial progenitor cells. Microvasc. Res. 78, 447–452 (2009).
Hoffmann, J. et al. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: Important role of nitric oxide. Circ. Res. 89, 709–715 (2001).
Widlansky, M. E., Gokce, N., Keaney, J. F. Jr. & Vita, J. A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 42, 1149–1160 (2003).
Rivard, A. et al. Age-dependent impairment of angiogenesis. Circulation 99, 111–120 (1999).
Bazzoni, G. & Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 84, 869–901 (2004).
Dejana, E., Tournier-Lasserve, E. & Weinstein, B. M. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev. Cell 16, 209–221 (2009).
Dudek, S. M. & Garcia, J. G. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl Physiol. (1985) 91, 1487–1500 (2001).
Gavard, J. & Gutkind, J. S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 8, 1223–1234 (2006).
Tiruppathi, C., Minshall, R. D., Paria, B. C., Vogel, S. M. & Malik, A. B. Role of Ca2+ signaling in the regulation of endothelial permeability. Vasc. Pharm. 39, 173–185 (2002).
Adams, R. H. & Eichmann, A. Axon guidance molecules in vascular patterning. Cold Spring Harb. Perspect. Biol. 2, a001875 (2010).
Treps, L., Le Guelte, A. & Gavard, J. Emerging roles of Semaphorins in the regulation of epithelial and endothelial junctions. Tissue Barriers 1, e23272 (2013).
Zhang, H. et al. Endothelial Semaphorin 3F maintains endothelial barrier function and inhibits monocyte migration. Int. J. Mol. Sci. 21. https://doi.org/10.3390/ijms21041471 (2020).
Kreivi, J. P. et al. Purification and characterisation of p99, a nuclear modulator of protein phosphatase 1 activity. FEBS Lett. 420, 57–62 (1997).
Landsverk, H. B. et al. The protein phosphatase 1 regulator PNUTS is a new component of the DNA damage response. EMBO Rep. 11, 868–875 (2010).
Boon, R. A. et al. MicroRNA-34a regulates cardiac ageing and function. Nature 495, 107–110 (2013).
Kim, Y. M. et al. PNUTS, a protein phosphatase 1 (PP1) nuclear targeting subunit. Characterization of its PP1- and RNA-binding domains and regulation by phosphorylation. J. Biol. Chem. 278, 13819–13828 (2003).
Kavela, S. et al. PNUTS functions as a proto-oncogene by sequestering PTEN. Cancer Res. 73, 205–214 (2013).
Kim, H. et al. TRF2 functions as a protein hub and regulates telomere maintenance by recognizing specific peptide motifs. Nat. Struct. Mol. Biol. 16, 372–379 (2009).
Ciurciu, A. et al. PNUTS/PP1 regulates RNAPII-mediated gene expression and is necessary for developmental growth. PLoS Genet 9, e1003885 (2013).
Dingar, D. et al. MYC dephosphorylation by the PP1/PNUTS phosphatase complex regulates chromatin binding and protein stability. Nat. Commun. 9, 3502 (2018).
De Leon, G., Sherry, T. C. & Krucher, N. A. Reduced expression of PNUTS leads to activation of Rb-phosphatase and caspase-mediated apoptosis. Cancer Biol. Ther. 7, 833–841 (2008).
Cortazar, M. A. et al. Control of RNA Pol II Speed by PNUTS-PP1 and Spt5 dephosphorylation facilitates termination by a “Sitting Duck Torpedo” mechanism. Mol. Cell https://doi.org/10.1016/j.molcel.2019.09.031 (2019).
Bukeirat, M. et al. MiR-34a regulates blood-brain barrier permeability and mitochondrial function by targeting cytochrome c. J. Cereb. Blood Flow. Metab. 36, 387–392 (2016).
Zhao, W. et al. MiR-34a regulates blood-tumor barrier function by targeting protein kinase Cepsilon. Mol. Biol. Cell 26, 1786–1796 (2015).
Michalik, K. M. et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ. Res 114, 1389–1397 (2014).
Hergenreider, E. et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat. Cell Biol. 14, 249–256 (2012).
Stanicek, L. et al. Long non-coding RNA LASSIE regulates shear stress sensing and endothelial barrier function. Commun. Biol. 3, 265 (2020).
Bonauer, A. et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 324, 1710–1713 (2009).
Wesseling, S. et al. Perinatal exogenous nitric oxide in fawn-hooded hypertensive rats reduces renal ribosomal biogenesis in early life. Front Genet 2, 52 (2011).
Wang, Y. et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486 (2010).
Hofmann, P. et al. Long non-coding RNA H19 regulates endothelial cell aging via inhibition of STAT3 signalling. Cardiovasc Res 115, 230–242 (2019).
Radu, M. & Chernoff, J. An in vivo assay to test blood vessel permeability. J. Vis. Exp., e50062. https://doi.org/10.3791/50062 (2013).
Yi, S. et al. NMR-based metabonomic analysis of HUVEC cells during replicative senescence. Aging (Albany NY) 12, 3626–3646 (2020).
Freedman, D. A. & Folkman, J. CDK2 translational down-regulation during endothelial senescence. Exp. Cell Res. 307, 118–130 (2005).
Wilhelm, K. et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 529, 216–220 (2016).
Varshavsky, A. et al. Semaphorin-3B is an angiogenesis inhibitor that is inactivated by furin-like pro-protein convertases. Cancer Res. 68, 6922–6931 (2008).
Mitsuhashi, S. et al. Tautomycetin is a novel and specific inhibitor of serine/threonine protein phosphatase type 1, PP1. Biochem. Biophys. Res Commun. 287, 328–331 (2001).
Choy, M. S. et al. PP1:Tautomycetin complex reveals a path toward the development of PP1-specific inhibitors. J. Am. Chem. Soc. 139, 17703–17706 (2017).
Zacharchenko, T., Barsukov, I., Rigden, D. J., Bennett, D. & Mayans, O. Biophysical analysis of the N-terminal domain from the human protein phosphatase 1 nuclear targeting subunit PNUTS suggests an extended transcription factor TFIIS-like fold. Protein J. 35, 340–345 (2016).
Fisher, L. A., Wang, L., Wu, L. & Peng, A. Phosphatase 1 nuclear targeting subunit is an essential regulator of M-phase entry, maintenance, and exit. J. Biol. Chem. 289, 23745–23752 (2014).
Grelet, S. et al. A regulated PNUTS mRNA to lncRNA splice switch mediates EMT and tumour progression. Nat. Cell Biol. 19, 1105–1115 (2017).
Yang, A. C. et al. Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature 583, 425–430 (2020).
Csortos, C., Kolosova, I. & Verin, A. D. Regulation of vascular endothelial cell barrier function and cytoskeleton structure by protein phosphatases of the PPP family. Am. J. Physiol. Lung Cell Mol. Physiol. 293, L843–L854 (2007).
Yazdani, U. & Terman, J. R. The semaphorins. Genome Biol. 7, 211 (2006).
Zhou, Y. et al. Reversal of gene dysregulation in cultured cytotrophoblasts reveals possible causes of preeclampsia. J. Clin. Invest 123, 2862–2872 (2013).
Sabag, A. D. et al. The role of the plexin-A2 receptor in Sema3A and Sema3B signal transduction. J. Cell Sci. 127, 5240–5252 (2014).
Acknowledgements
This work was supported by the German Center for Cardiovascular Research (DZHK), the Deutsche Forschungsgemeinschaft (SFB834 to R.A.B. and S.D.), the Netherlands Organisation for Scientific Research (NWO Vidi to R.A.B.), and the European Union (Horizon 2020 Grant No. 825670 to R.A.B.).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
N.L.-V. designed and performed experiments, analyzed data and drafted and revised the paper; L.S., D.I.B., R.P.J., A.H., V.K., P.P. and A.vB. performed experiments, analyzed data and revised the paper; A.W.M. performed experiments and analyzed data; S.D. provided conceptual input and revised the paper; R.A.B. supervised the project, designed experiments, analyzed data, and drafted and revised the paper.
Corresponding author
Ethics declarations
Competing interests
N.L.-V. and L.S. are currently employed by AstraZeneca. All other authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Patric Turowski and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Joao Valente.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lozano-Vidal, N., Stanicek, L., Bink, D.I. et al. Aging-regulated PNUTS maintains endothelial barrier function via SEMA3B suppression. Commun Biol 7, 541 (2024). https://doi.org/10.1038/s42003-024-06230-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06230-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
