
Abstract
The hemibiotrophic Basidiomycete pathogen Ganoderma boninense (Gb) is the dominant causal agent of oil palm basal stem rot disease. Here, we report a complete chromosomal genome map of Gb using a combination of short-read Illumina and long-read Pacific Biosciences (PacBio) sequencing platforms combined with chromatin conformation capture data from the Chicago and Hi-C platforms. The genome was 55.87 Mb in length and assembled to a high contiguity (N50: 304.34 kb) of 12 chromosomes built from 112 scaffolds, with a total of only 4.34 Mb (~ 7.77%) remaining unplaced. The final assemblies were evaluated for completeness of the genome by using Benchmarking Universal Single Copy Orthologs (BUSCO) v4.1.4, and based on 4464 total BUSCO polyporales group searches, the assemblies yielded 4264 (95.52%) of the conserved orthologs as complete and only a few fragmented BUSCO of 42 (0.94%) as well as a missing BUSCO of 158 (3.53%). Genome annotation predicted a total of 21,074 coding genes, with a GC content ratio of 59.2%. The genome features were analyzed with different databases, which revealed 2471 Gene Ontology/GO (11.72%), 5418 KEGG (Kyoto Encyclopedia of Genes and Genomes) Orthologous/KO (25.71%), 13,913 Cluster of Orthologous Groups of proteins/COG (66.02%), 60 ABC transporter (0.28%), 1049 Carbohydrate-Active Enzymes/CAZy (4.98%), 4005 pathogen–host interactions/PHI (19%), and 515 fungal transcription factor/FTFD (2.44%) genes. The results obtained in this study provide deep insight for further studies in the future.
Similar content being viewed by others
Introduction
The oil palm of African origin, Elaeis guineensis, is the most productive oil-bearing crop in the world. This species was introduced by the Dutch into Indonesia in 1848 as an ornamental palm at the Bogor Botanical Garden1. It has become a fundamental oil palm ancestor of the current massive cultivation of this plant species in Southeast Asia, particularly Indonesia and Malaysia. This oil crop produces an average of 3.5 tons of oil per hectare, although it has a full yield potential of up to 18 tons/ha/year from the third year onward2. Moreover, with its current status, palm oil contributes almost one-third (75.17 million tons) of the world’s total vegetable oils3. The oil benefits the food, oleochemical, and renewable energy source sectors4. However, the industry is facing serious yield limitations due to basal stem rot (BSR) disease.
BSR is associated with 15 Ganoderma spp. (Basidiomycota: Polyporales) strains worldwide5. However, pathogenic interactions with oil palm have been reported for only three species, i.e., G. boninense (Gb), G. zonatum, and G. miniatocinctum6,7. Although Gb is a dominant pathogen for BSR, the latter two species have rarely been reported as causal agents of BSR cases in the field8,9 (Fig. 1). Nevertheless, BSR remains a major economically important problem in the oil palm industry in Indonesia, Malaysia and Papua New Guinea10,11,12. Despite deep interest in fungi, there is little genetic and genomic information available to provide insights into host-interactions13,14,15. On the other hand, a complete genome sequence of another Ganoderma fungus, G. lucidum (Gl), revealed remarkable gene cooperation for secondary metabolite biosynthesis as well as the richest sets of wood degradation enzymes among all sequenced basidiomycetes16. Interestingly, Gl has been famous for its edible pharmaceutical herb value for thousands of years17,18, rather than being a hemibiotroph phytopathogen, as shown by Gb19,20. Therefore, in this study, a genome comparison analysis might provide insight into the evolutionary histories, scenes and trajectories of both fungi, which impart speciation and lifestyle.

BSR incidence in the field showing fruiting bodies of Gb emerging from the basal stem of an oil palm tree.
Recent advances in sequencing technologies have made the full genome assembly of interesting species relatively affordable. At least eight commercial sequencing platforms are available, despite each having their own merits, whereas one platform could be better fit for a study than another depending on the purpose21. In general, these platforms are based mainly on short- and long-read sequencing approaches in which the divergences are related to the throughput, cost, error rate, and read structure22. The combination of short- and long-read sequencing approaches, such as the Illumina HiSeq 4000 and Pacific Bioscience platforms, was used to generate a high-quality whole-genome sequence and assembly of Auricularia heimuer (Basidiomycota: Auriculariales) and Arthrinium phaeospermum (Ascomycota: Sordariomycetidae)23,24. Moreover, the birth of the 3C (chromosome conformation capture) technique in 2002 involving formaldehyde crosslinking of nuclei provides pairwise linking information between reads that originate from genomic regions that are physically adjacent in a nucleus25. The combination of Illumina and PacBio sequencing coupled with the two most common 3C derivative protocols, Chicago and Hi-C, allows the collection of fragments/bins/genomic loci that are profiled as read-pair interactions on an “all-by-all” basis in the entire genome26,27,28. In addition, the HiRise™ software pipeline builds an accurate and contiguous genome assembly, identifies poor-quality joins and produces accurate, long-range sequence scaffolds29. Proximity-based ligation and massive sequencing of reads form a three-dimensional architecture of chromosomes (Hi-C) with a high probability of intrachromosomal contacts, even when separated by > 200 Mb, to generate a reasonably accurate chromosome-scale de novo assembly30,31. Here, we report the de novo genome assembly of Gb using Illumina short reads and PacBio long reads coupled with the Chicago + HiRise assembler to construct scaffolds, and the resulting scaffolds were subsequently aligned to the Hi-C + HiRise platform to generate chromosome levels.
Results
Global genome assembly and characteristics
The genome sequence of the Gb strain G3 was used in this study32,33. A fine draft genome sequence was constructed by the hybrid assembly of short reads on the Illumina HiSeq 4000 platform combined with long reads on the PacBio RS II platform to generate a draft genome of this fungus. The libraries from both platforms were assembled using WTDBG234, followed by short- and long-read iterative correction using each two-round Racon and Pilon as the polishers constitutively35,36. The hybrid sequences from the two platforms were assembled de novo to construct a 55.82 Mb total draft genome sequence with 826x sequencing coverage, containing 592 genomic scaffolds with an N50 length of 0.357 Mb.
In the downstream scaffolding, the contigs were scaffolded in a series of HiRise analyses, initially using the Chicago data followed by inclusion of the Hi-C reads for chromosome-scale assembly of Gb. Using the Chicago approach, the library produced 165 million reads of 2 × 150 bp or provided 1887.11× physical coverage of the genome. The Hi-C library produced 194 million read pairs of 2 × 150 bp or provided 41,650.61× physical coverage of the genome. Using Illumina and PacBio reads as input assembly data, the Chicago HiRise pipeline for assembly resulted in a 55.86 Mb total assembled sequence with 264 scaffolds. This result was used as an input assembly for the HiRise assembler and resulted in a final 112 scaffolds with a total genome assembly length of 55.87 Mb (Table 1). The scaffold joined by the Hi-C mate pairs introduced new “N” gaps in the assembly, thereby increasing the number of gaps in the assembly to 7201, and each gap consisted of 100 “N”. The final genome assembly was characterized by 21,074 coding genes, with a GC content ratio of 59.2%. The average gene length was 1265 bp, while the average intergenic size was 1250 bp. The average number of exons per gene was 7, with average exon and intron sizes of 178 and 97 bp, respectively (Table 2).
The contiguity comparison graph showed a significant improvement in the final assembly (Fig. 2A), and the library insert size distribution graph exhibited an accurate distribution of reads to facilitate the repeat bridging and resolution capabilities of the state-of-the-art assembly (Fig. 2B). In addition, the HiRise pipeline generated a Hi-C linkage density histogram (Fig. 2C). The alignment produces a diagonal of lines from the lower left to upper right in the plot that represent each of the 12 Gb chromosome-scale scaffolds ranging from 3.03 to 6.65 Mb (Fig. 2D, Table S1).

Chromosome assembly of Gb via dovetail genomics. (A) Comparison of the contiguity of the input assembly and the final HiRise scaffolds. Each curve shows the fraction of the total length of the assembly present in scaffolds of a given length or shorter. The fraction of the assembly is indicated on the Y-axis, and the scaffold length in base pairs is given on the X-axis. The two dashed lines mark the N50 and N90 lengths of each assembly. (B) The distribution of insert sizes. The distance between the forward and reverse reads is given on the X-axis in base pairs, and the probability of observing a read pair with a given insert size is shown on the Y-axis. (C) Hi-C linkage density histogram. In this figure, the x- and y-axes show the mapping positions of the first and second reads in the read pair, respectively, which are grouped into bins. The color of each square indicates the number of read pairs within that bin. Dots (sequences) within boxes in the last column are probably 4.34 Mb of unassembled scaffold DNA sequence with only ~ 7.77% bases. White vertical and black horizontal lines have been added to show the borders between the scaffolds. (D) Chromosome structures illustrating the gene density on each chromosome.
The final assemblies were evaluated for genome completeness by using BUSCO37 version 4.1.4 based on 4464 total BUSCO polyporales group searches. The assemblies yielded 4264 (95.52%) of the conserved orthologs as complete common eukaryote gene sequence coverage, with only a few missing (158, or 3.53%), as well as a duplicate score of 90 (2.01%) and a fragmented BUSCO of 42 (0.94%). In addition, BUSCO analysis of Agaricomycetes and Basidiomycota group searches was also conducted, and the results were similar. The completeness levels were 93.16% and 93.53% for the two groups, respectively (Fig. 3, Table S2). This result indicates that our genome assembly is of relatively high quality and contiguous.

Three BUSCO scores of the Polyporales, Agaricomycetes, and Basidiomycota group searches illustrating the assembly and annotation completeness of the Gb genome.
Genome assembly could be an issue in diploid or polyploid samples due to possible discrepancies within reads or contigs that arbitrarily alternate between parental alleles for certain genomic regions. Indeed, our assembly revealed the extent of natural heterozygosity in the diploid Gb mycelial samples used for sequencing. De novo assemblies were used to merge homologous and heterozygous loci into single “consensus” sequences. Variant analysis in this study detected 527,941 high-confidence polymorphisms for the entire genome by aligning sequence reads to the total assembled Gb genome and calling variants, including insertions-deletions (indels) and nucleotide polymorphisms (SNPs). We found, on average, 9 variants in every 1 kb of the Gb genome (Table S3). However, in this study, the BUSCO analysis results demonstrated the intact de novo assembly of Gb from a diploid material genetic source.
Gene prediction and functional annotation
Among the 21,074 annotated genes in the Gb genome, we characterized their functions using different databases, including the Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthologous (KO), Cluster of Orthologous Groups of Protein (COG), ATP-Binding Cassette (ABC) Transporter, Carbohydrate-Active Enzyme (CAZy), Pathogen-Host Interaction (PHI), and Fungal Transcription Factor (FTFD) databases. GO analysis has become the most complete, comprehensive, and wide-ranging method for predicting gene function in various eukaryotic species. In this study, the GO database annotated 2471 coding genes in Gb categorized into three major classes, i.e., biological process, cellular component, and molecular function. Biological process was the most enriched function, with 25,648 annotations, followed by cellular component and molecular function, with 12,494 and 9976 annotations, respectively (Table S4). Most of the biological processes were annotated as proteolysis (GO:0006508), with 108 (0.42%) annotations, while most of the cellular component and molecular function terms were related to protein binding (GO:0005515) and cytosol (GO:0005829), with 591 (5.66%) and 841 (4.92%) annotations, respectively.
Subsequently, 25.71% (5418) of the Gb coding sequences were annotated via the KO database. The biological pathways are divided into nine KO categories, each of which is subdivided, and each category is labeled with the relevant information. Among these predicted genes, those involved in the metabolism function category accounted for the majority (30.58%, 1657) of the annotated genes. In contrast, those categorized as “others” were the least annotated at 0.79% (43) (Fig. 4, Table S5). We also enriched all the coding sequences of the assembled Gb genome with the COG database. The functional analyses revealed that 66.02% (13,913 genes) of the coding sequences were annotated. However, almost half (6524) of the annotations were categorized as poorly categorized. This might be because the database was built from predominantly unicellular microorganisms of 1187 bacterial and 122 archaeal genomes and only seven sequenced eukaryotic genomes, including those of Homo sapiens, Drosophila melanogaster, Caenorhabditis elegans, Arabidopsis thaliana, Saccharomyces cerevisiae, Schizosaccharomyces pombe, and Encephalitozoon cuniculi38,39. Moreover, those with known functions are relatively common in all living organisms, and their abundance is equally distributed among the three COG categories, such as information storage and processing, cellular process and signaling, and metabolism (Fig. 5 top, Table S6).

The KO functional annotation of the Gb genome is shown. The major classes and the number of genes from the subclass partitions of interest are represented. The X-axis represents the scale for the number of genes. KEGG functional annotation was divided into nine major classes: cellular process, environmental information processing, genetic information processing, human diseases, metabolism or organismal system, and 40 subclasses. Each class is represented by a distinct color.

COG functional classification of proteins in the Gb genome. The X-axis represents the scale for the number of genes. COG functional annotation is divided into four classes: information storage and processing, cellular process and signaling, metabolism, and poorly characterized. Each class is represented by a distinct color.
In other analyses, we also identified the presence of ABC transporter proteins in the Gb genome that actively transport a wide substrate repertoire and range of functions. A total of 60 predicted genes were found, which were distributed in eight families, whereas the full-length multidrug resistance protein (MRP/ABC-C) family contributed the most genes (Fig. 6-left, Table S7). Additionally, CAZy, a carbohydrate-degrading enzyme, is a dominant virulence factor in white-rot Basidiomycetes. Here, our CAZy prediction revealed 1049 encoding genes in the Gb genome, accounting for 4.98% of all annotated genes. Among the classes, glycoside hydrolases (GHs) were the most common, with 385 genes (37%), followed by genes involved in auxiliary activities (AAs), glycosyl transferases (GTs), carbohydrate esterases (CEs), carbohydrate-binding modules (CBMs), and polysaccharide lyases (PLs) (Fig. 6-right, Table S8).

The distribution of ABC transporters and CAZy families in the Gb genome. Left. The ABC family parentheses indicate structural component domains in each ABC family. B. The abundance of CAZys for each family in the genome.
Moreover, we found a total of 4005 predicted Gb genes associated with pathogen-host interactions (PHIs). Among those, 4613 were annotated as the highest proportion, as high as 48.58% (2241), which was related to reduced virulence, followed by unaffected pathogenicity (29.37%), loss of pathogenicity (9.91%), increased virulence (5.66%), lethality (5.53%), and others below 1% (Fig. 7A, Table S9). Transcription factors (TFs) are important regulators of gene expression for modulating diverse biological processes, and some have been reported to act as virulence factors for fungal phytopathogens40. In this study, we reported that 515 genes encoding TFs were identified in the Gb, accounting for 2.84% of the total number of predicted genes. The largest class was the zinc finger CCHC (pfam domain: PF00098.26), with 169 members or 32.82% of the total TFs. This is followed by fungal trans (pfam domain: PF04082.21) and zinc finger C2H2 (pfam domain: PF00096.29), with 70 and 65 members, or 13.59% and 12.62%, respectively. The other classes were distributed at 10% and below (Fig. 7B, Table S10).

The abundance of predicted PHI phenotypic-category genes and distribution of transcription factor families in Gb. The percentage of each PHI category (A) and TF family (B) relative to the total number of predicted genes.
Discussion
Ganoderma Karst. (Ganodermataceae, Polyporales) is a cosmopolitan genus of white-rot fungi with more than 200 members41,42. Above, we reported high-quality chromosomal genome sequence assemblies of Gb, a white-rot fungal member that plays an ecological niche and is an economically important plant pathogen1,10,43, whereas BUSCO analysis supported its completeness and accuracy in ensuring the reliability of the gene structures37. The genome capacity and number of Gb genes are slightly greater than the average of all available Basidiomycota fungal whole-genome information, i.e., 55.87 out of 46.48 Mb and 21,074 out of 15,431 genes, respectively44. The available genome size of Basidiomycota fungi ranges from 9.82 (Wallemia sebi) to 130.65 Mb (Dendrothele bispora), even though the latest genome sequence project for some edible mushrooms has expanded in size to 202.2 Mb45. The revolution of sequencing and genome assembly technology paves the way for genomics studies of species of interest to reveal their indispensable genomic structure. Chromosome conformation capture techniques, such as the Hi-C and Dovetail Genomics Chicago libraries, in combination with long-read library sequencing platforms help span and resolve repetitive regions, thus greatly improving genome assembly46. In particular, Hi-C sequencing is considered an economical method for generating chromosome-scale scaffolds and for genome reconstruction47. The Hi-C assay is based on in vivo proximity ligation, along with the following principles: (1) Hi-C interactions tend to occur within the same chromosome because chromosomes occupy distinct territories and spatial preferences in the nucleus. (2) Hi-C separates nuclei after proximity ligation because genomic DNA is fixed within an intact nucleus, and cross-linking between nuclei is rare. (3) The Hi‒C interaction frequency exhibits genomic distance-dependent decay because according to the principles of polymer physics, cis-loci in linear sequences interact more frequently than trans-loci48. It is thus reliable to use Hi-C data to determine that contigs with frequent interactions are likely to belong to the same chromosome31,49,50.
In principle, we generated sufficient PacBio sequencing coverage to improve the scaffold N50 length, polished the assembly with Illumina short reads, and further used data from Dovetail Genomics Chicago and Hi‐C libraries to extend the scaffolds to the chromosomal level. The availability of long-read PacBio sequencing technology paves the way for achieving longer contigs with better quality assemblies, especially for the sequencing of large repeat regions that are not properly assembled or missed using short-read-based methods. Long-read sequencing coupled with Hi-C library technology is currently one of the best approaches for generating high-quality chromosome levels without knowledge of chromosome numbers. Dovetail™ Hi-C data can provide links across a variety of length scales, spanning even whole chromosomes, and this technique has been used to improve draft genome assemblies and create chromosome-length scaffolds for large genomes. The Dovetail Chicago and Hi‐C libraries increased the longest scaffold by more than threefold, from 1.6 to 6.6 Mbp or one-eighth of the total Gb genome, and the scaffold N50 increased by more than 11‐fold, from 0.36 to 4.13 Mbp, which means that the contiguity significantly improved. Another metric for assessing the quality of assemblies is the completeness of the so-called BUSCO. A “good” genome assembly criterion is expected to contain approximately 90% of the BUSCO gene set51. In this assessment, the BUSCO completeness scores of 98.56%, 93.53%, and 93.16% for Polyporales, Basidiomycota and Agaricomycetes, respectively, indicate that the current assembly is of high quality and highly consistent. In the future, other techniques, including optical mapping from BioNano52 combined with PacBio, may further improve the assembly quality, even though the joint accuracy of BioNano is reported to be 15% greater in Chicago53. In this approach, the methodology described here makes it feasible to obtain a near-finished or completely assembled Gb genome, which is reflected in the ability to obtain 12 scaffolds that correspond to 12 chromosomes in Gb.
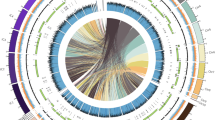
Ganoderma Karst. (Ganodermataceae, Polyporales) is a cosmopolitan genus of white rot fungi with more than 200 members41,42. The two best-known taxa economically play ecological niches either as plant pathogens for various tree crops (Gb) or as commercial edible herbal medicines (Gl)54. Previously, three whole-genome sequence projects of Ganoderma species were reported, each for Gl, Ganoderma sp., and G. sinense16,55,56. The first and last mentioned fungi are known for their medicinal properties, while Ganoderma sp. was described as a forest saprophyte. However, Gl is more popular and well characterized, providing us with the opportunity to appraise any genomic discrepancy that may underlie the different ecological niches and perhaps to identify pathogenicity factors in Gb. Moreover, the available high-quality genome assemblies of Gb and Gl, which are early-sequenced Ganoderma fungi, facilitate pairwise comparisons of genomic features and specific components (Fig. 8).

Circos plot of the genomic features of Gb versus Gl. The circles from outside to inside represent the chromosome map, sequence gaps (A), GC content (B), gene density (C), and CAZys content of the six families; AA (D1), CBM (D2), CE (D3), GH (D4), GT (D5), and PL (D6). Syntenic gene pairs are linked by colored lines in the inner-most circle. Each feature was calculated based on 100 kb nonoverlapping windows.
Both Gb and Gl are white-rot fungi that degrade plant cell walls via lignocellulose depolymerization57. A side-by-side comparison between both species revealed that the number of copies of CAZY-encoding genes in Gb was greater than that in Gl. Most likely, this study may describe a complex mechanism underpinning divergent ecological niches between Gb as a phytopathogen and Gl as a saprobe. A richer repertoire of CAZYs observed in Gb is likely needed to enable the degradation of living woody tissues of oil palm, whereas two-thirds of oil palm trunk biomass is composed of lignocellulose (17.1% lignin, 41.2% cellulose, and 34.4% hemicellulose)58. As commonly found in other fungi, both fungal species also contain the GH family, which has the largest number of CAZys, and many of the enzymes play a crucial role in the hydrolysis of carbon‒oxygen‒carbon bonds and hereinafter break sugar residues and utilize them for consumption59. Additionally, the complete set of ligninolytic enzymes comprises phenol oxidase CAZy AA1 (laccases: EC 1.10.3.2) and peroxidase CAZy AA2 (lignin peroxidase/EC 1.11.1.14; manganese peroxidase/EC 1.11.1.13; and versatile peroxidase/EC 1.11.1.16) to catalyze the oxidation of phenolic units in lignin, and free phenoxy radicals are present in both fungi60. This is because fungal laccases and peroxidases are involved in the depolymerization of lignin, defense or protection against antimicrobial effects, virulence and/or phytopathogenicity61.
Methods
Fungal strain
We used a Gb strain from our collection cultures labeled the G3 strain. Previously, this strain was used to generate a draft genome32. The fungus was maintained in presterilized rubber wood sawdust (RWS) culture in the dark at 28 °C and subcultured every 6 months. Approximately 10 mg of RWS was revived on potato dextrose agar (PDA) for 7 days each time prior to use. Freshly pure revived mycelia were grown in 100 ml of yeast malt broth in the dark at 28 °C for 14 days. Mycelia were harvested on a layer of Whatman paper no. 1 and air-dried for 15 min. Genomic DNA was processed using a GenElute Plant Genomic DNA Miniprep Kit (Sigma–Aldrich Co., St. Louis, MO, USA) according to the manufacturer’s instructions for PacBio and Illumina platform sequencing. Freeze-dried mycelia samples were shipped to Dovetail Genomic, Inc., USA, for Chicago and Hi-C library preparation.
Illumina/PacBio sequencing and de novo assembly
Single-molecule sequencing was performed using PacBio RS II with the latest P6-C4 chemistry systems and an Illumina HiSeq 4000 system according to each manufacturer’s instructions. The PacBio reads were assembled using WTDBG2 with default parameters34. Subsequently, two rounds of Racon were used for consensus calling for this assembly. Running a Racon for long reads, two or several rounds, can increase the accuracy of bridging areas between contigs, generate high-quality consensus sequences, and achieve further iterations of the consensus sequence35. At the final assembly step, two rounds of PacBio-Racon assemblies were polished by basecall correction through Pilon software with default parameters36. Pilon uses Illumina reads to perform base corrections and derive an accurate consensus sequence. Moreover, at least two rounds of pilon polishing are required to achieve the same level of accuracy in most cases62.
Chicago consolidant library preparation and sequencing
The Chicago library preparation and sequencing were outsourced to Dovetail Genomics (Chicago, USA) and performed as follows. Briefly, ~ 500 ng of high-molecular-weight genomic DNA (gDNA) (mean fragment length = 59) was reconstituted into chromatin in vitro and fixed with formaldehyde. The fixed chromatin was digested with DpnII, the 5’ overhangs were filled with biotinylated nucleotides, and the free blunt ends were subsequently ligated. After ligation, the crosslinks were reversed, and the DNA was purified from the protein. The purified DNA was treated to remove biotin that was not internal to the ligated fragments. The DNA was then sheared to an ~ 350 bp mean fragment size, and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina-compatible adapters. Biotin-containing fragments were isolated using streptavidin beads before PCR enrichment of each library. The libraries were sequenced on an Illumina HiSeq X platform to produce 165 million 2 × 150 bp paired-end reads, which provided 1887.11 × physical coverage of the genome (1–100 kb pairs).
Hi-C library preparation and sequencing
Hi-C library preparation and sequencing were also performed by Dovetail Genomics (Chicago, USA) as follows. Briefly, for each library, chromatin was fixed in place with formaldehyde in the nucleus and then extracted. The fixed chromatin was digested with DpnII, the 5ʹ overhangs were filled with biotinylated nucleotides, and the free blunt ends were subsequently ligated. After ligation, the crosslinks were reversed, and the DNA was purified from the protein. The purified DNA was treated to remove biotin that was not internal to the ligated fragments. The DNA was then sheared to an ~ 350 bp mean fragment size, and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina-compatible adapters. Biotin-containing fragments were isolated using streptavidin beads before PCR enrichment of each library. The libraries were sequenced on an Illumina HiSeq X platform to produce 194 million 2 × 150 bp paired-end reads, which provided 41,650.61× physical coverage of the genome (10–10,000 kb pairs).
Scaffolding the assembly with HiRise
The input de novo assembly, shotgun reads, Chicago library reads, and Dovetail Hi-C library reads were used as input data for HiRise, a software pipeline designed specifically for using proximity ligation data to scaffold genome assemblies29. An iterative analysis was conducted. First, the shotgun and Chicago library sequences were aligned to the draft input assembly using a modified SNAP read mapper (http://snap.cs.berkeley.edu). The separations of Chicago read pairs mapped within draft scaffolds were analyzed by HiRise to produce a likelihood model for genomic distance between read pairs, and the model was used to identify and break putative misjoins, to score prospective joins, and to make joins above a threshold. After aligning and scaffolding the Chicago data, Dovetail Hi‒C library sequences were aligned and scaffolded following the same method. After scaffolding, the shotgun sequences were used to close gaps between contigs.
Gene annotation
Protein-coding gene prediction was performed using the de novo prediction tool BRAKER with the default parameters59. It is a novel automated protein-coding gene predictor program that works through a combination of GeneMark-ET60 for protein-coding gene prediction and is used as a training set for the AUGUSTUS pipeline61. GeneMark-ET was subjected to RNA-seq read alignment to assemble the RNA-seq transcripts of Gb derived from in vitro growth in rich potato dextrose broth media for 14 days. This transcript evidence was confirmed by reference to the available reference protein of G. sinense52.
All the predicted Gb transcripts were annotated against the UniProtKB/Swiss-Prot database using BLASTX software with a cutoff of an E-value restriction of 10–634. The annotations of the sequences with the best scores were chosen to be the annotations of the genes according to the functional term classification of each database. The GO enrichment analysis was performed using the enricher function from the PANNZER, while KO, COG, and PHI analyses were each performed using TRAPID 2.0, EggNOG 5.0, and PHI-base according to their default parameters62,63,64,65. The annotations for ABC transporters, CAZys, and transcription factors were performed using HMMer (version 3.0) based on each particular motif in the PANTHER database66.
Variants and calling SNPs and short INDELs
The Illumina reads were aligned to the reference genome of Gb using BWA-MEM63. Variants with a minimum quality > 30 were called using BCFtools (mpileup and call)64. Variants with a depth < 20 reads, minor allele frequency (MAF) < 0.05, missing data < 10%, and number of alleles > 2 were removed using VCFtools65.
Genome comparison analysis
Syntenic genes between Gb and Gl were identified using LASTZ66. Comparative genome analysis was carried out by comparing genome characteristics, including CAZys, which are characteristic of white-rot fungi, with the previously reported genome sequence of Gl16. All putative Gb proteins were searched against entries in the CAZy database (http://www.cazy.org/) using BLASTP. The proteins with e-values smaller than E-06 were further screened by a combination of BLAST searches against individual protein domains belonging to the AA, CBM, CE, GH, GT, and PL class motifs. HMMer (version 3.0) was used to query against a collection of custom-made hidden Markov model (HMM) profiles constructed for each CAZy class. All identified proteins were then manually curated.
Ethics declaration
All the authors declare that plant collection and use were in accordance with all the relevant guidelines.
Plant material permission
All authors state appropriate permissions and/or licenses for collection of plant or seed specimens used in this study.
Data availability
The whole-genome sequence data of this study has been deposited at DDBJ/EMBL/GenBank (http://www.ncbi.nlm.nih.gov/) under the accession no. PJEW03000000 version.
References
Corley, R. H. V. & Tinker, P. B. The Oil Palm (Blackwell Science Ltd, 2016).
Barcelos, E. et al. Oil palm natural diversity and the potential for yield improvement. Front. Plant Sci. 6, 1–16 (2015).
Kushairi, A. et al. Oil palm economic performance in Malaysia and r&d progress in 2017. J. Oil Palm Res. 30, 163–195 (2018).
Maluin, F. N., Hussein, M. Z. & Idris, A. S. An overview of the oil palm industry: Challenges and some emerging opportunities for nanotechnology development. Agronomy 10, 356 (2020).
Turner, D. P. Oil Palm Diseases and Disorders. Incorporated Society of Planters (Oxford University Press, 1981).
Idris, A. S., Ariffin, D., Swinburne, T. R. & Watt, T. A. The identity of Ganoderma species responsible for BSR disease of oil palm in Malaysia. MPOB Information series 4 (2000).
Wong, L. C., Bong, C. F. J. & Idris, A. S. Ganoderma species associated with basal stem rot disease of oil palm. Am. J. Appl. Sci. 9, 879–885 (2012).
Rakib, M. R. M., Bong, C. F. J., Khairulmazmi, A. & Idris, A. S. Aggressiveness of Ganoderma boninense and G. zonatum isolated from upper - and Basal stem rot of oil palm (Elaeis guineensis) in Malaysia. J. Oil Palm Res. 27, 229–240 (2015).
Rakib, M. R. M., Khairulmazmi, A., Idris, A. S., Jalloh, M. B. & Wahida, N. H. Ganoderma species of basal and upper stem rots in oil palm (Elaeis Guineensis) in Sarawak. J. Acad. UiTM Negeri Sembilan 5, 27–35 (2017).
Aderungboye, F. O. Diseases of the oil palm. Pans 23, 305–326 (1977).
Pilotti, C. A., Sanderson, F. R. & Aitken, E. A. B. Genetic structure of a population of Ganoderma boninense on oil palm. Plant Pathol. 52, 455–463 (2003).
Pilotti, C. A., Sanderson, F. R., Aitken, E. A. B. & Armstrong, W. Morphological variation and host range of two Ganoderma species from Papua New Guinea. Mycopathologia 158, 251–265 (2004).
Mercière, M. et al. Identification and development of new polymorphic microsatellite markers using genome assembly for Ganoderma boninense, causal agent of oil palm basal stem rot disease. Mycol. Prog. https://doi.org/10.1007/s11557-015-1123-2 (2015).
Voo, C. L. Y., Yeo, D. E. T., Chong, K. & Rodrigues, K. F. Draft genome sequence of a phytopathogenic Ganoderma sp. Microbiol. Resour. Announc. 8–9 (2020).
Sulaiman, S., Othman, N. Q., Tan, J. S. & Lee, Y. P. Draft genome assembly dataset of the Basidiomycete pathogenic fungus, Ganoderma boninense. Data Br. 29, 105167 (2020).
Chen, S. et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 3, 913–919 (2012).
Loyd, A. L. et al. Identifying the ‘Mushroom of immortality’: Assessing the Ganoderma species composition in commercial reishi products. Front. Microbiol. 9, 1–14 (2018).
Stamets, P. Growing Gourmet and Medicinal Mushrooms (Ten Speed Press, 1993).
Rees, R. W., Flood, J., Hasan, Y., Potter, U. & Cooper, R. M. Basal stem rot of oil palm (Elaeis guineensis); Mode of root infection and lower stem invasion by Ganoderma boninense. Plant Pathol. 58, 982–989 (2009).
Bahari, M. N. A., Sakeh, N. M., Abdullah, S. N. A., Ramli, R. R. & Kadkhodaei, S. Transciptome profiling at early infection of Elaeis guineensis by Ganoderma boninense provides novel insights on fungal transition from biotrophic to necrotrophic phase. BMC Plant Biol. 18, 1–25 (2018).
Mardis, E. R. DNA sequencing technologies: 2006–2016. Nat. Protoc. 12, 213–218 (2017).
Goodwin, S., McPherson, J. D. & McCombie, W. R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351 (2016).
Yuan, Y., Wu, F., Si, J., Zhao, Y. F. & Dai, Y. C. Whole genome sequence of Auricularia heimuer (Basidiomycota, Fungi), the third most important cultivated mushroom worldwide. Genomics 111, 50–58 (2019).
Li, S. et al. Whole-genome sequence of Arthrinium phaeospermum, a globally distributed pathogenic fungus. Genomics 112, 919–929 (2020).
Dekker, J., Rippe, K., Dekker, M. & Kleckner, N. Capturing chromosome conformation. Science 295, 1306–1311 (2002).
Ghurye, J. & Pop, M. Modern technologies and algorithms for scaffolding assembled genomes. PLoS Comput. Biol. 15, 1–20 (2019).
Oluwadare, O., Highsmith, M. & Cheng, J. An overview of methods for reconstructing 3-D chromosome and genome structures from Hi-C data. Biol. Proced. Online 21, 1–20 (2019).
Cairns, J. et al. CHiCAGO: Robust detection of DNA looping interactions in capture Hi-C data. Genome Biol. 17, 1–17 (2016).
Putnam, N. H. et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26, 342–350 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Utomo, C. et al. Draft genome sequence of the phytopathogenic fungus Ganoderma boninense, the causal agent of basal stem rot disease on oil palm. Genome Announc. 6, 1–2 (2018).
Utomo, C. et al. High quality genome sequence reveals the 12 pseudo-chromosomes of Ganoderma boninense. Bio Rxiv 1–8. https://doi.org/10.1101/817510(2019).
Ruan, J. & Li, H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2020).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Walker, B. J. et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, 1–14 (2014).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Tatusov, R. L. et al. The COG database: An updated vesion includes eukaryotes. BMC Bioinform. 4, 1–14 (2003).
Galperin, M. Y. et al. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 49, D274–D281 (2021).
John, E., Singh, K. B., Oliver, R. P. & Tan, K. C. Transcription factor control of virulence in phytopathogenic fungi. Mol. Plant Pathol. 22, 858–881 (2021).
Richter, C., Wittstein, K., Kirk, P. M. & Stadler, M. An assessment of the taxonomy and chemotaxonomy of Ganoderma. Fungal Divers. 71, 1–15 (2015).
Du, Z., Dong, C. H., Wang, K. & Yao, Y. J. Classification, Biological Characteristics and Cultivations of Ganoderma. Advances in Experimental Medicine and Biology vol. 1181 (2019).
Pilotti, C. A. Stem rots of oil palm caused by Ganoderma boninense: Pathogen biology and epidemiology. Mycopathologia 159, 129–137 (2005).
Mohanta, T. K. & Bae, H. The diversity of fungal genome. Biol. Proced. Online 17, 1–9 (2015).
Li, H. et al. The Genome Sequences of 90 Mushrooms. Sci. Rep. 8, 6–10 (2018).
Elbers, J. P. et al. Improving Illumina assemblies with Hi-C and long reads: An example with the North African dromedary. Mol. Ecol. Resour. 19, 1015–1026 (2019).
Ghurye, J. et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comput. Biol. 15, 1–19 (2019).
Xu, Z. & Dixon, J. R. Genome reconstruction and haplotype phasing using chromosome conformation capture methodologies. Brief. Funct. Genomics 19, 139–150 (2020).
Lajoie, B. R., Dekker, J. & Kaplan, N. The Hitchhiker’s guide to Hi-C analysis : Practical guidelines. Methods 72, 65–75 (2016).
Kaplan, N. & Dekker, J. High-throughput genome scaffolding from in vivo DNA interaction frequency. Nat. Biotechnol. 31, 1143–1147 (2013).
Hanschen, E. R., Hovde, B. T. & Starkenburg, S. R. An evaluation of methodology to determine algal genome completeness. Algal Res. 51, 102019 (2020).
Nagarajan, N., Read, T. D. & Pop, M. Scaffolding and validation of bacterial genome assemblies using optical restriction maps. Bioinformatics 24, 1229–1235 (2008).
Moll, K. M. et al. Strategies for optimizing BioNano and Dovetail explored through a second reference quality assembly for the legume model, Medicago truncatula. BMC Genomics 18, 1–16 (2017).
Flood, J., Bridge, P. D. & Hodlerness, M. Ganoderma Diseases of Perennial Crops (CABI Publishing, 2000). https://doi.org/10.1079/9780851993881.0000.
Binder, M. et al. Phylogenetic and phylogenomic overview of the Polyporales. Mycologia 105, 1350–1373 (2013).
Zhu, Y. et al. Chromosome-level genome map provides insights into diverse defense mechanisms in the medicinal fungus Ganoderma sinense. Sci. Rep. 5, 1–14 (2015).
Paterson, R. R. M. Ganoderma disease of oil palm—A white rot perspective necessary for integrated control. Crop Prot. 26, 1369–1376 (2007).
Paterson, R. R. M., Moen, S. & Lima, N. The feasibility of producing oil palm with altered lignin content to control Ganoderma disease. J. Phytopathol. 157, 649–656 (2009).
Murphy, C., Powlowski, J., Wu, M., Butler, G. & Tsang, A. Curation of characterized glycoside hydrolases of Fungal origin. Database 2011, 1–14 (2011).
Andlar, M. et al. Lignocellulose degradation: An overview of fungi and fungal enzymes involved in lignocellulose degradation. Eng. Life Sci. 18, 768–778 (2018).
Janusz, G. et al. Laccase properties, physiological functions, and evolution. Int. J. Mol. Sci. 21, 1–25 (2020).
Chen, Z., Erickson, D. L. & Meng, J. Polishing the Oxford Nanopore long-read assemblies of bacterial pathogens with Illumina short reads to improve genomic analyses. Genomics 113, 1366–1377 (2021).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 00, 1–3 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Harris, R. S. Improved Pariwise Alignment of Genomic DNA (Pennsylvania State University, 2007). https://doi.org/10.1016/j.jocs.2018.04.005.
Acknowledgements
We thank the Management of PT. SMART Tbk financially supported this project. We thank Sanju Rianintika and Hani Feorani for their technical support. We also thank Roberdi, Yudistira Wahyu Kurnia, and Victor Aprilyanto for their suggestions, corrections, and proofreading to improve this manuscript.
Author information
Authors and Affiliations
Contributions
CU and TL led and supervised the project. RA, ZAT and HSGP performed the bioinformatics and statistical analyses. RFNB and ADMP perform fungal cultivation and artificial infection in planta. RT conceived the study and wrote the manuscript. All authors approved the submitted manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Utomo, C., Tanjung, Z.A., Aditama, R. et al. Whole-genome sequencing of Ganoderma boninense, the causal agent of basal stem rot disease in oil palm, via combined short- and long-read sequencing. Sci Rep 14, 10520 (2024). https://doi.org/10.1038/s41598-024-60713-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-60713-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
