
Abstract
In digenic inheritance, pathogenic variants in two genes must be inherited together to cause disease. Only very few examples of digenic inheritance have been described in the neuromuscular disease field. Here we show that predicted deleterious variants in SRPK3, encoding the X-linked serine/argenine protein kinase 3, lead to a progressive early onset skeletal muscle myopathy only when in combination with heterozygous variants in the TTN gene. The co-occurrence of predicted deleterious SRPK3/TTN variants was not seen among 76,702 healthy male individuals, and statistical modeling strongly supported digenic inheritance as the best-fitting model. Furthermore, double-mutant zebrafish (srpk3−/−; ttn.1+/−) replicated the myopathic phenotype and showed myofibrillar disorganization. Transcriptome data suggest that the interaction of srpk3 and ttn.1 in zebrafish occurs at a post-transcriptional level. We propose that digenic inheritance of deleterious changes impacting both the protein kinase SRPK3 and the giant muscle protein titin causes a skeletal myopathy and might serve as a model for other genetic diseases.
Similar content being viewed by others

Main
Over the past decade, next-generation sequencing (NGS) has contributed greatly to diagnostics in rare diseases. Nevertheless, many individuals considered to be affected by a genetic condition remain undiagnosed1. One underlying reason for this may be that the prevailing diagnostic paradigm still adheres to the one-gene one-disease model. Although thousands of monogenic diseases have been described, true digenic inheritance, where deleterious variants in two independent genes must be present for the disease to manifest, is scarce2. Here we describe a cohort of individuals with a skeletal muscle myopathy (henceforth referred to as myopathy) caused by co-inheritance of deleterious variants that impact both the muscle-specific protein kinase serine/arginine protein kinase 3 (SRPK3) and the giant muscle protein titin.
Titin, encoded by TTN, is the largest known protein and is expressed in cardiac and skeletal muscles. Spanning the Z-disk to the M-band, it is involved in sarcomeric assembly and function3. Expression and processing of TTN is age- and tissue-specific and involves complex transcriptional regulation4,5. Pathogenic variants in TTN cause a range of skeletal and cardiac phenotypes, which are inherited mostly in recessive6,7,8,9,10 and dominant forms11, respectively. However, due to its sheer size and extensive alternative splicing, interrogation and interpretation of genetic variants and protein expression data is challenging12.
SRPK3 encodes a protein kinase member of the SRPK family that phosphorylates proteins containing serine-arginine dipeptide motifs (SR proteins)13. In humans, three tissue-specific SRPKs have been described14,15, with SRPK3 being expressed predominantly in striated muscle16. SRPKs primarily regulate both constitutive and alternative mRNA splicing through the phosphorylation of SR-splicing factors and spliceosomal components13,17. SRPK3 is essential for muscle growth and homeostasis13,14,16, and skeletal muscle is highly sensitive to SRPK3 expression levels, as not only its deficiency but also its overexpression led to an abnormal muscle phenotype in mice, reminiscent of a human centronuclear myopathy16. In view of the mouse data, we initially considered SRPK3 to be a myopathy candidate gene, but our subsequent findings support a more complex model.
Results
SRPK3 variants alone do not explain disease manifestation
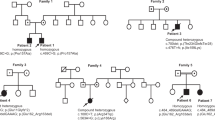
In our cohort of 2,170 exome datasets from patients with neuromuscular disease, we identified five males (of 1,170) hemizygous for deleterious variants in the X-linked SRPK3 gene. Through international collaborations, we expanded the collection to a total of 33 patients with myopathy (31 males and two females, from 25 families) carrying deleterious variants in SRPK3. The majority (64%) were high-impact variants (stop gain, frameshift and splicing; Table 1). RNA analysis of splice variants showed abnormal mRNA splicing (Extended Data Fig. 1), expected to lead to nonsense-mediated decay, and SRPK3 mRNA counts per million (CPM) in three individuals with truncating variants was significantly lower than controls (−1.981 log fold change, P = 7.106 × 10−11; Extended Data Fig. 2). All missense variants were located in one of SRPK3’s kinase domains (Extended Data Fig. 3a) and were predicted to be deleterious (Table 1). 3D modeling anticipated restriction of the backbone conformation or disruption of the helical structure, causing instability and reduced catalytic activity (Extended Data Fig. 3b). All but one of the SRPK3 variants were absent among 76,702 control males from gnomAD (v2.1.1; https://gnomad.broadinstitute.org/)18. The two manifesting female carriers in family Y (YIII:4 and YIII:5) showed skewed X-inactivation in lymphocytes, which could explain their phenotypes. Segregation analyses (in 20 families) showed that SRPK3 variants were inherited from an unaffected mother, with none being de novo, and were present in all affected male siblings. However, SRPK3 variants did not cosegregate with the disease as nine unaffected males from seven families (KII:2, LII:2, QII:1, VII:2/3/4, WIII:1, YIV:1 and ZIII:2) also carried the familial SRPK3 variants (Fig. 1a and Supplementary Table 1).

a, Segregation of the familial SRPK3 variants is shown. S indicates the SRPK3 variant and WT indicates the wild-type allele. Individuals presenting with skeletal muscle disease are indicated in black. Mild presentations are shown in gray (corresponding to YIII:4 and YIII:5, two female carriers with skewed X-inactivation, 80:20 and 65:35, in lymphocytes, respectively). b, Extended pedigree details of families M and Z. Individuals presenting with skeletal muscle disease are indicated in black. Cardiac involvement is indicated by gray/dotted symbols. Segregation of the familial SRPK3 (S) and TTN (T) variants is shown. S + T indicates cosegregating SRPK3/TTN variants; WT indicates both SRPK3 and TTN WT alleles. Individuals ZIV:1, ZIV:4, ZIV:6 and ZIV:7 carry the familial TTN variant (p.Arg16905*) previously reported in association with DCM (ref. 19) but are presymptomatic at ages 52, 44, 40 and 38 years old, respectively. Likewise, individual MIII:2 carries the familial TTN variant (p.Asp28805Metfs*6) but is also presymptomatic at age 46 years old. c, Cosegregation of the SRPK3 and TTN variants (S + T) with the myopathic phenotype (shown in black). All known genotypes are shown; WT, both SRPK3 and TTN WT alleles; empty symbols indicate that the sample was not available for testing (or failed testing). All affected individuals carry the SRPK3 and TTN variants (S + T), whereas their unaffected relatives carry one or the other, but never both. Two females carrying cosegregating SRPK3/TTN variants and showing a skewed X-inactivation pattern are mildly affected (YIII:4 and YIII:5), and those with random X-inactivation are unaffected (TI:2, UI:2, XII:2 and YII:2). A female carrying only the SRPK3 variant (but no TTN variant; ZII:5) and a complete X-inactivation pattern (3:97, in lymphocytes) is unaffected. Individual RI:2, with cosegregating SRPK3 and TTN variants whose fully inactivated chr X carries the SRPK3 deleterious variant, is also unaffected. Individuals RII:3 and SI:2 are noninformative for the CAG repeat analyzed in the X-inactivation assay.
We then noted that, in two of our extended SRPK3 pedigrees (families M and Z; Fig. 1b), some family members presented with isolated dilated cardiomyopathy (DCM). In both families, the DCM was associated with a dominantly inherited heterozygous truncating variant in TTN —a new frameshift variant in exon 326 (p.Asp28805Metfs*6) in family M, and a stop-gain variant (p.Arg16095*), previously reported in association with DCM19, in family Z. In these families, the patients with myopathy also presented with DCM and carried the familial ‘cardiac’ TTN variant. Interestingly, the myopathic phenotype (in patients MIII:3, ZIII:4, ZIII:7 and ZIV:1) only manifested when the SRPK3 variant was present in combination with the TTN variant (Fig. 1b and Supplementary Table 1).
Taking this into account, we reassessed our myopathy families and screened them for TTN variants. All the index cases, in addition to the SRPK3 variants, carried a heterozygous variant in the TTN gene. The vast majority (84%) were TTN truncating variants (TTNtv), and all were absent, or extremely rare, in the control population (Table 1). No variant clustering was observed (Extended Data Fig. 4). Segregation analyses showed that the myopathy manifested only if both the SRPK3 and TTN variants were inherited together, but not when either variant was present in isolation (Fig. 1c). This was also the case for the two manifesting female carriers in family Y (YIII:4 and YIII:5), as they too carried a new deleterious heterozygous TTN variant. In contrast, females with cosegregating SRPK3 and TTN variants but who had random X-inactivation were unaffected (that is, TI:2, UI:2, XII:2 and YII:2). The exception to this was RI:2, an unaffected 72-year-old female with cosegregating SRPK3 and TTN variants whose fully inactivated chromosome (chr) X was confirmed to carry the SRPK3 deleterious allele. Also unaffected was individual ZII:5, a female SRPK3 carrier showing a fully skewed X-inactivation pattern (3:97) but no TTN variant (Fig. 1c and Supplementary Table 1).
SRPK3/TTN cases present with a slowly progressive myopathy
Individuals with cosegregating SPRK3/TTN variants presented with a relatively homogenous phenotype and clinical course. Disease onset was in childhood or earlier (30/33), with poor motor performance. The disease was slowly progressive (23/33), yet all but four patients were ambulatory at the last assessment. The mean age of patients was 32 years (range: 1–77 years). The pattern of weakness was predominantly proximal and axial, affecting the lower more than the upper limbs. Respiratory compromise was present in 14 individuals, four of whom required noninvasive nocturnal ventilation. Three patients had DCM; however, this could be attributed to their TTNtv11. All patients had normal or mildly elevated serum creatine kinase (CK) levels, except RII:1, who had consistently elevated CK values (2,400:U/l). Less frequent features and deep-phenotype descriptions are listed in Supplementary Tables 2 and 3. Histopathology for 23 skeletal muscle biopsies showed myopathic changes with increased internalized nuclei (22/23), core-like structures (15/23) and type I fiber predominance (19/23). Electron microscopy (EM) images confirmed the presence of core-like areas and revealed myofibrillar disorganization with Z-line streaming and branching of myofibrils (Fig. 2). Axial T1-weighted muscle magnetic resonance imaging (MRI) scans of the lower limbs in four patients showed a similar selective pattern of muscle pathology with prominent fatty transformation of the subscapularis, gluteus maximus, adductor longus, vasti, hamstrings, medial gastrocnemius and soleus muscles. The sartorius, gracilis and adductor magnus muscles were well preserved (Fig. 2).

a–h, Examples of muscle histopathology (n = 23). a, Myopathic changes with increased internalized nuclei and fiber size variability (22/23) shown by hematoxylin and eosin (H&E) staining, as seen in patient XIII:1. b,c, Minicores and core-like structures (15/23) shown by NADH histochemistry, as seen in patients BII:1 and WIII:3. d,e, Type I fiber predominance and type I uniformity (19/23) shown by ATPase pH 4.6 and pH 9.2 staining, as seen in patients CII:2 and BII:1, respectively. f, More severe end of the disease spectrum, with vacuoles, necrosis, regeneration and fibrosis shown by H&E in patient YII:3. g,h, EM images confirmed the presence of core structures and revealed Z-line misalignment, accumulation of Z-band material and branching of myofibrils, as seen in patients XIII:1 and WIII:3. Representative images have been obtained as part of the diagnostic workup in accredited pathology laboratories. i, Lower limb MRI T1-weighted images from four patients with SRPK3/TTN myopathy (VII:1, YII:3, MIII:3 and ZIV:1). A pattern of fatty replacement involving the subscapularis muscle in the shoulder girdle was observed. In the pelvic girdle, the gluteus maximus was affected (arrows), but the gluteus minimus and medius muscles were spared even in the advanced stages of the disease. In the thigh, there was a predominant involvement of the hamstring muscles, while the sartorius and gracilis muscles were not involved in the advanced stages of the disease, with the adductor magnus muscle (arrowheads) almost completely spared. In the lower legs, there was predominant involvement of the medial gastrocnemius muscle (arrows) associated with the involvement of the soleus muscle. The peroneus and tibialis anterior muscles were also involved, but only in advanced stages.
Abnormal titin expression in patients with SRPK3/TTN myopathy
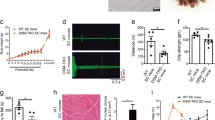
Based on the available genetic data, no evident copy number variants (CNVs) or variants in the triplicated region of the TTN gene were found in trans with the heterozygous TTN variant. However, to further exclude compound heterozygosity for a TTN defect abolishing the titin C-terminal, we used western blotting to detect the small C-terminal titin proteolytic fragments (13, 15 and 18 kDa in size)20. Muscle biopsy protein lysates of patients DII:1, LII:1, XIII:1 and YII:3 showed a normal titin C-terminal pattern (at least from one allele), ruling out a biallelic C-terminal titinopathy (Fig. 3a). Antibodies against the N-terminus and distal I-band of titin showed that the full-length titin band was present in a TTNtv carrier (DI:1) but appeared to be missing or reduced in the patients with SRPK3/TTN myopathy (CII:2, XIII:1 and YII:3), suggesting that the normal full-length expression of both TTN alleles was affected (Fig. 3b,c). In keeping with this, transcriptome analysis of patients LII:1, DII:1 and YII:3 showed that TTN mRNA expression (as length-normalized CPM) was significantly lower than controls (−1.450 log fold change, P = 8.015 × 10−4; Extended Data Fig. 5).

Muscle biopsy lysates of individuals DI:1, DII:1, LII:1, XII:3, XIII:1 and YII:3 were analyzed using different anti-titin antibodies. a, SRPK3/TTN patients LII:1, YII:3, DII:1, XII:3 and XIII:1 showed a normal pattern of C-terminal titin proteolytic fragments (13, 15 and 18 kDa in size), ruling out a C-terminal titinopathy. b,c, Antibodies against the N-terminal titin (Z1Z2 TTN-1, b) and distal I-band of titin (F146.9B9, c) showed that the full-length titin band is missing or highly reduced in the patients with SRPK3/TTN myopathy (XIII:1, XII:3 and YII:3), but it is present in an unaffected relative TTNtv carrier (DI:1, father of DII:1) and a disease control also carrying a heterozygous TTNtv. This could be attributed to changes in N-terminal protein sequence or structure, or otherwise, protein modifications preventing antibody recognition. d, Coomassie staining also showed the absence or reduction of the high molecular weight band representing the full-length titin protein, whereas the NEB and MyHC bands were normal. Western blots were repeated twice, from the same muscle lysates. Full-length blots are provided as source data. MW, molecular weight; MyHC, myosin heavy chain; NEB, nebulin.
TTN truncating variants are enriched in the SRPK3 cohort
To elucidate whether the co-occurrence of SRPK3 and TTN variants observed in our patients could be due to chance, we compared our findings with both control and other disease populations. We focused exclusively on TTNtv, as TTN missense variants are too abundant and would pose a challenge for correct pathogenicity ascertainment. First, we interrogated the gnomAD database and estimated that TTNtv were present at ~1% in the control population, in keeping with previous reports21. Next, we analyzed three cohorts of genetically confirmed limb-girdle muscular dystrophies: LGMD-R1 (n = 170), LGMD-R2 (n = 94) and LGMD-R12 (n = 56). While 21 of the 25 SRPK3 families carried a TTNtv (84%), we found five heterozygous TTNtv in the patients with LGMD-R1 (2.94%; P = 6.13 × 10−19), one in the patients with LGMD-R2 (1.06%; P = 9.18 × 10−17) and two in the patients with LGMD-R12 (3.5%, P = 4.89 × 10−12; Extended Data Fig. 6). We then queried the existence of healthy male individuals carrying high-impact variants in both SRPK3 and TTN in the control population of exomes from gnomAD. As of September 2023, there are six hemizygous males (of 76,702) carrying five truncating variants in SRPK3 canonical transcript (p.Ser30Ter, p.Lys139GlnfsTer10, c.475+1G>A, p.Arg373Ter and p.Lys516SerfsTer17; ENSG00000184343; https://gnomad.broadinstitute.org/gene). Manual interrogation of the exome data of these six males disclosed that none of them carried a TTNtv, highlighting that co-occurring truncating hemizygous SRPK3 and heterozygous TTN variants, as seen in our patients, is a very unusual event (P = 0.00232; Supplementary Note). Finally, we used statistical modeling to quantify the degree to which our observations supported the co-inheritance of causal SRPK3 and TTN variants as opposed to any other plausible explanation. The best-fitting model involves digenic inheritance, with both SRPK3 and TTN variants required for disease manifestation. The likelihood of data is at least 1010 times greater than under any other model, including a model where just one gene (that is, SRPK3 or TTN) is operating, but with reduced penetrance (Supplementary Note).
Zebrafish double mutants replicate the SRPK3/TTN human myopathy
We next tested whether our observations for SRPK3 and TTN could be replicated in an animal model using srpk3 and ttn zebrafish mutant lines—the srpk3sa18907 mutation causes aberrant splicing, leading to partial retention of intron 15 or loss of exon 15 (Extended Data Fig. 7); ttn.1sa5562 is a premature stop codon in exon 19 of 214 of the ttn.1 gene. Zebrafish have two paralogs for the human TTN gene, ttn.1 and ttn.2, with ttn.1 exclusively affecting skeletal muscle function22 (Extended Data Fig. 8). We created double carrier zebrafish of srpk3sa18907 and ttn.1sa5562 (srpk3+/sa18907; ttn.1+/sa5562, henceforth referred to as srpk3+/−; ttn.1+/−) and used sibling in-crosses to produce offspring carrying all possible genotype combinations, including srpk3−/−; ttn.1+/− (henceforth referred to as double mutant). Zebrafish single (srpk3+/+; ttn.1+/−) and double (srpk3+/−; ttn.1+/−) heterozygous mutant larvae at 5 dpf (days postfertilization; Fig. 4b,f) had no phenotype and were indistinguishable from the wild-type (WT; Fig. 4a,e). Double-mutant zebrafish (srpk3−/−; ttn.1+/−) at first appeared to be morphologically normal (including the heart), but they were not able to fill the swim bladder and therefore did not survive to adulthood. Compared to WT fish (Fig. 4a,e), the muscle fiber structure was largely lost in the ttn-null fish (both srpk3+/+; ttn.1−/− and srpk3−/−; ttn.1−/−; Extended Data Fig. 8), but was only very mildly affected in the srpk3-null fish (srpk3−/−; ttn.1+/+; Fig. 4c, g). In accordance with the findings in our patients with myopathy, however, the loss of one ttn.1 WT allele in the srpk3-null zebrafish resulted in severe muscle pathology (srpk3−/−; ttn.1+/−; Fig. 4d,h), as visualized by whole mount staining of actin filaments and Z-band markers. Although myotomes were properly formed, muscle fibers were distorted and disintegrated to a variable extent. EM of the mutant zebrafish showed that the sarcomere appeared unaffected in heterozygous ttn.1 (srpk3+/+; ttn.1+/−; Fig. 3r), comparable to WT zebrafish (Fig. 4q). The srpk3-null zebrafish (srpk3−/−; ttn.1+/+; Fig. 4s) showed mildly disorganized myofibrils; however, most sarcomeres appeared well defined. The double-mutant fish (srpk3−/−; ttn.1+/−; Fig. 4t) displayed pronounced disruption of the sarcomere structure, including the myofibrils, A-band, I-band, H-zone and M-line. For further characterization, we isolated zebrafish myofibers and immunostained them with a monoclonal anti-titin antibody. This antibody labeled the T11 peptide found at the I-band to A-band transition. We observed, by confocal imaging, that srpk3−/−; ttn.1+/− double mutants (Fig. 4l,p) developed disorganized sarcomeres compared both to ttn.1 heterozygous (srpk3+/+; ttn.1+/−; Fig. 4j,n) and srpk3-null mutants (srpk3−/−; ttn.1+/+; Fig. 4k,o). This disruption of the myofibers was also illustrated by a substantial reduction of titin labeling and disturbance of the transverse labeling pattern of α-actinin. These results were consistent with the I-band and A-band alterations observed in the EM images.

a–h, Lateral view of Alexa Fluor phalloidin filamentous actin (green) and α-actinin Z-band marker (red) staining in skeletal fast muscle fibers in WT (a,e), srpk3+/−; ttn.1+/− (b,f), srpk3−/−; ttn.1+/+ (c,g) and srpk3−/−; ttn.1+/− larvae (d,h) at 5 dpf. Compared to WT (a,e) or double heterozygotes (srpk3+/−; ttn.1+/−; b,f), homozygous srpk3-null alone only causes very mild muscle fiber defects (c,g), while ttn.1 heterozygosity in homozygous srpk3−/− larvae severely affects muscle fiber integrity (d,h). i–t, Isolated myofiber immunostaining and electron microscopy (EM) in skeletal fast muscle fibers in WT (i,m,q), srpk3+/+; ttn.1+/− (j,n,r), srpk3−/−; ttn.1+/+ (k,o,s) and srpk3−/−; ttn.1+/− (l,p,t) larvae at 5 dpf. Isolated myofiber immunostaining showed that titin expression is largely reduced in the double mutant (srpk3−/−; ttn.1+/−; l,p) but not in the single heterozygous ttn.1 mutant (srpk3+/+; ttn.1+/−; j,n) or the srpk3-null (srpk3−/−; ttn.1+/+; k,o). EM showed that srpk3-null zebrafish (srpk3−/−; ttn.1+/+; s) had well-defined sarcomeres, with mildly disorganized myofibrils. The double-mutant fish (srpk3−/−; ttn.1+/−; t) displayed pronounced disruption of the sarcomere structure. White scale bars are 25 µm. Black scale bar is 500 nm. Representative images from >15 pooled fish per genotype.
Transcriptome analysis highlights disruption of contractile structures in zebrafish double mutants
Zebrafish transcriptome data showed that ttn.1 mRNA expression is equally reduced in the heterozygous (srpk3+/+; ttn.1+/−) and the double mutants (srpk3−/−; ttn.1+/−) when compared to WT ttn.1 (Extended Data Fig. 9a). This suggests that the severe reduction of titin protein expression observed by immunostaining exclusively in the double mutants (srpk3−/−; ttn.1+/−; Fig. 4l) was due to aberrant post-transcriptional or post-translational processing. In contrast, srpk3 mRNA levels were upregulated in srpk3−/− mutants (both srpk3−/−; ttn.1+/+ and srpk3−/−; ttn.1+/−; Extended Data Fig. 9b), likely as a compensatory effect. When analyzing the global transcriptome profile of the different genotypes, we observed that there were 128 genes differentially expressed (DE) in the heterozygous ttn (srpk3+/+; ttn.1+/−) zebrafish. The number of DE genes in the srpk3-null mutant (srpk3−/−; ttn.1+/+) was 572, and this increased to 794 in the double mutant (srpk3−/−; ttn.1+/−; Fig. 5a). A Gene Ontology (GO) enrichment analysis to identify the differential pathways involved showed that the transcriptional changes in srpk3-null (srpk3−/−; ttn.1+/+) zebrafish and the double mutant (srpk3−/−; ttn.1+/) were similar. Expression of genes involved in myofibril and actin cytoskeletal organization and skeletal muscle tissue development was affected. There was also an inflammatory signal in both genotypes (Fig. 5b,c, Extended Data Fig. 10 and Supplementary Data). Unexpectedly, the heterozygous ttn.1 mutant (srpk3+/+; ttn.1+/−), despite being fully viable and having no morphological phenotype, also showed a clear inflammatory signature, but no dysregulation of muscle genes (Fig. 4b and Supplementary Data). In addition, given the role of SRPK3 in RNA processing and maturation, we queried for generalized aberrant splicing patterns, but we did not detect any signal for this.

a, Number of DE genes between WT and double mutant (top: srpk3−/−; ttn.1+/−; n = 794), srpk3-null (middle: srpk3−/−; ttn.1+/+; n = 572) and heterozygous ttn.1 (bottom: srpk3+/+; ttn.1+/−; n = 128) zebrafish. Upregulated genes are in blue and downregulated genes are in red. b, GO term enrichment analysis. GO term enrichment was done using the topGO package using a one-sided Fisher’s exact test without adjustment for multiple testing. The top enriched GO terms (P < 0.001) for the three comparisons in a ordered by −log10(P). The bars are colored according to the GO domain. Blue indicates BP; orange indicates CC; green indicates MF. c, ClueGO network diagram showing the overlap in enriched GO terms between double mutant (srpk3−/−; ttn.1+/−) and srpk3-null (srpk3−/−; ttn.1+/+). Nodes represent individual enriched GO terms; edges connect nodes that share annotated genes from the DE genes. Nodes are colored according to the contribution to the enrichment from DE genes from each comparison. Blue indicates >60% DE genes from the srpk3−/−; ttn.1+/− comparison; red indicates >60% DE genes from srpk3−/−; ttn.1+/+; purple indicates 40–60% from each comparison. BP, biological process; CC, cellular component; MF, molecular function.
Ttn variants in Srpk3 knockout (KO) mouse
We then interrogated the genetic background of the Srpk3 KO mouse described in ref. 16 to establish whether its resulting muscle phenotype was also due to the cosegregation of Srpk3 and a previously unrevealed Ttn variant. Using genome sequencing, we found the following three Ttn changes in the Srpk3 KO mouse model: two missense (chr2:76946873C>T; p.Ala1395Val and chr2:76969682C>T; p.Ala394Val) and one synonymous variant (chr2:76969699T>C; p.Ser388Ser), also present in the WT 129s6/SvEvTAC background (http://www.informatics.jax.org/snp/). We currently do not know whether these variants contribute to the observed phenotype.
SRPK3 in vitro phosphorylates RNA-binding motif 20 (RBM20), a splicing factor involved in TTN mRNA regulation
Protein expression analysis of muscle biopsy lysates from our patients with SRPK3/TTN myopathy suggested that SRPK3 deficiency might affect normal full-length titin expression. This was later supported by the reduction in titin labeling seen in the zebrafish double-mutant (srpk3−/−; ttn.1+/−) model. SRPK3 could be directly involved in titin phosphorylation or more likely, given the regulatory role of serine/arginine (SR) kinases, in TTN mRNA processing by targeting an SR-splicing regulator. RBM20 is a muscle-specific splicing factor involved in TTN alternative splicing23. Its cellular localization and activity are dependent on the phosphorylation of its RSRSP stretch within the arginine/serine-rich region24. We hypothesized that RBM20 might be a phosphorylation substrate of SRPK3 and the mediating link between SRPK3 and titin, as previously shown for SRPK1 (ref. 25). To investigate this, we cotransfected an RBM20 reporter (RBM20517–664-V5) into 293T cells with or without a GFP-SRPK3 construct. The presence of GFP-SRPK3 led to RBM20517–664-V5 hyperphosphorylation, as indicated by a mobility shift of the reporter (Fig. 6a). This mobility shift was abolished after treatment with lambda phosphatase. This suggests that SRPK3 may directly phosphorylate the TTN-specific splicing factor, RBM20. Based on this finding, we interrogated the transcriptome data for the zebrafish mutants and found that the zebrafish rbm20 ortholog shows increased expression upon loss of srpk3 (both srpk3−/−; ttn.1+/+ and srpk3−/−; ttn.1+/−; Fig. 6b).

a, The RBM20517–664-V5 reporter was transfected into 293T cells with or without GFP-SRPK3. GFP-SRPK3/RBM20517–664-V5 co-expression resulted in RBM20517–664-V5 hyperphosphorylation (lanes 4 and 5), as indicated by a mobility shift that was abolished by incubation with lambda phosphatase (P, lane 6). U indicates untreated samples; N indicates control samples incubated without phosphatase. In the absence of the SRPK3 construct, a less pronounced but still noticeable mobility shift can be observed (lanes 1 and 2), consistent with RBM20 phosphorylation by endogenous kinases such as SRPK1, CLK1 or AKT2. Assay was performed in quadruplicate. b, mRNA counts of the zebrafish RBM20 ortholog (BX649294.1 ENSDARG00000092881) are increased in srpk3-null zebrafish (srpk3−/−; ttn.1+/+ and srpk3−/−; ttn.1+/−), likely as a feedback loop due to the srpk3 deficiency. The box blots represent the first and third quartiles (25% and 75% percentile) with the center line at the median value. The whiskers extend from the hinge to the furthest value not beyond 1.5 times the interquartile range from the hinge. Differential expression was done using a two-sided Wald test with Benjamini–Hochberg adjustment for multiple testing63. For srpk3−/−; ttn.1+/+ versus srpk3+/+; ttn.1+/+, *P = 0.0379. n = 6 for each condition. Full-length blots are provided as source data.
Discussion
We identified 40 males (from 25 families) carrying hemizygous deleterious variants in the X-linked SRPK3 gene. Of those, only the 31 patients who also carried cosegregating heterozygous TTN variants presented with a myopathy. Their unaffected brothers carried either the SRPK3 or the TTN variant, but never both. For the female individuals, a mild presentation was observed only in the two sisters from family Y who carried both the TTN and the X-linked SRPK3 variants and showed skewed X-inactivation. However, a female SRPK3 carrier displaying skewed X-inactivation, but no TTN variant, was unaffected. The remaining females with SRPK3/TTN variants but random X-inactivation were also unaffected. While numbers are small, this might suggest that, for SRPK3 carrier females to present a myopathic phenotype, both skewed X-inactivation and a deleterious TTN variant must co-occur, in line with what is observed in male patients.
Disease and control population data indicated that the co-occurrence of SRPK3 and TTN variants was not fortuitous, because TTNtv variants were significantly more common in patients with SRPK3/TTN myopathy than in other genetically diagnosed muscular dystrophy cohorts, and were notably absent in the ‘SRPK3-null’ males present in the control population. These findings, together with the statistical modeling, strongly support the digenic inheritance of deleterious SRPK3 and TTN variants in patients with myopathy. While digenic inheritance has been widely recognized in association with, for example, deafness26,27 and cardiovascular conditions28, only a handful of digenic cases have been reported in the neuromuscular field. These are, however, mostly single cases29,30, with only SQSTM1/TIA1 multisystem proteinopathy (MPS)31 and D4Z4/SMCHD1 facioscapulohumeral muscular dystrophy type 2 (FSHD2; ref. 32) being replicated in independent cohorts. To our knowledge, this is the first report of true digenic inheritance involving a protein kinase in a sizeable cohort of skeletal myopathy patients.
The zebrafish model, where the srpk3−/−; ttn.1+/− double-mutant embryos showed a severe muscle phenotype not observed in the srpk3−/− or ttn.1+/− embryos alone, replicated our findings. In addition, the model allowed us to better understand the muscle pathology, highlighting the disorganization of the sarcomere and the reduction in titin protein expression. Transcriptome analysis showed that compared to WT, ttn.1 mRNA expression levels were similarly reduced in the single ttn.1+/− heterozygous mutant, both with or without srpk3-null background, suggesting that post-transcriptional (or post-translational) processing must be responsible for the abnormally expressed protein. Likewise, differential gene expression analysis demonstrated that, despite severe morphological consequences, losing one ttn.1 WT allele in srpk3-null mutants only had a minor effect on the gene expression profile. This suggests that the interaction of srpk3 and ttn.1 in zebrafish is at a post-transcriptional level.
The in vitro phosphorylation assay supported a connection between SRPK3 and the TTN-splicing factor RBM20. Increased mRNA counts of the rbm20 zebrafish ortholog were observed exclusively in the srpk3-null mutants, possibly the result of a positive feedback loop due to srpk3-related phosphorylation deficiency of rbm20. Similar upregulation was seen in a knock-in RBM20 mouse model (Rbm20S637A) where phosphorylation was impaired33. While in our hands the srpk3-null 5-dpf zebrafish appeared normal with almost unaffected fiber structure, at the time of submission an adult KO model was shown to present agenesis of cerebellar structures and abnormal behavior34, suggesting srpk3 may also be involved in neural development.
Most of the TTN variants identified in our SRPK3 cohort were truncating, yet novel missense variants, predicted deleterious, were also found. While more challenging to ascertain35, TTN missense changes have been shown to be disease-causing in homozygosity or compound heterozygosity with a TTNtv or other missense change35,36,37. In heterozygosity, in particular in exons 344 and 364, TTN missense changes are associated with dominant hereditary myopathy with early respiratory failure (HMERF)10,11 and tibial muscular dystrophy (TMD)7,9. Three of the TTNtv variants seen in our cohort were previously reported. Two of these (p.Arg16095* and p.Arg31056*) had been associated with DCM (ref. 19 and Leiden Open Variation Database, https://databases.lovd.nl/shared/genes/TTN) and were identified in two (of the three) patients with SRPK3/TTN myopathy also presenting with DCM (families B and Z). The third variant (p.Arg8494*) had been reported in a patient with an unsolved muscle disease38 who was analyzed through a panel of 35 neuromuscular disease genes. Given our findings, it would be appropriate to screen the SRPK3 gene in such unsolved patients with sporadic myopathy and a heterozygous TTNtv.
Notably, none of the family members who carried only the heterozygous TTN variant (n = 16) showed any signs of skeletal muscle disease, in keeping with what has been largely accepted for heterozygous TTNtv6,8,20. Notwithstanding, it has been reported recently that heterozygous TTNtv in the A-band may be causative of dominant distal myopathy39. When no evident dominant family history exists, however, it would be worth considering whether a more complex molecular pathomechanism might be responsible for these presentations.
N-terminal blots of a TTNtv carrier showed expression of full-length titin, yet when similar variants were present in combination with SRPK3 variants, only smaller or weaker bands seemed to be detected. Similarly, reduction of titin immunolabeling was observed in the heterozygous ttn.1 zebrafish model but only in an srpk3-null background. The epitope for the anti-titin antibody is located in region T11 (around exon 102), downstream of the premature stop codon generated by the ttn.1sa5562 mutation (chr9:42861631T>G, exon 19); therefore, only WT titin would have been detected by immunostaining. This suggests that the loss of SRPK3 negatively affects the WT TTN copy, either by directly altering protein structure or conformation, or more likely, by post-transcriptional processing (possibly through RBM20 regulation), resulting in loss of antibody recognition. We propose that the myopathy observed in the patients with SRPK3/TTN myopathy, and replicated in the zebrafish model, is the result of a titin dosage effect, whereby a single ‘faulty copy’ of TTN is not sufficient to cause disease, but the additional deficiency in SRPK3 activity, affecting TTN transcriptional regulation and, in turn, normal full-length titin expression, tilts the scale toward pathology.
We have shown that, in vitro, SRPK3 phosphorylates at least one of the serine residues present in the transfected RBM20517–664-V5 construct, corresponding to the RNA-recognition motif and RS-rich domains and including the RSRSP stretch. RSRSP phosphorylation is critical for RBM20 localization and activity24,33. RBM20 is a muscle-specific SR-splicing factor, primarily involved in I-band TTN alternative splicing, and known to regulate the ratio of N2BA:N2B cardiac isoforms40. Additional splicing targets include other sarcomeric genes (for example, OBSCN and LDB3), genes essential for calcium handling (for example, CAMKD2 and RYR2) and even neuronal regulation (for example, SEMA6D)41. Mutations in RBM20 have been associated with highly penetrant and severe dominant DCM in humans and other mammals41,42. Most frequently, these are gain-of-function missense changes in the highly conserved RSRPS region leading to cytoplasmic retention and aberrant ribonucleoprotein granules24,43,44. Conversely, loss-of-function (LoF) mutations outside the RSRSP stretch result in RBM20 haploinsufficiency and aberrant splicing of target genes, such as TTN, but not mislocalization45,46. This is in line with population data showing RBM20 to be highly LoF intolerant (pLI = 0.99)18. We manually interrogated the exome data of healthy RBM20 LoF carriers and, interestingly, no cosegregating TTNtv were identified, just as seen with the SRPK3 LoF hemizygotes.
It has been postulated that RBM20-DCM is more severe than TTN-DCM and thus cannot be solely explained by aberrant TTN-splicing regulation47. Notably, all but three of the SRPK3/TTN families did not present cardiac involvement. Speculatively, abnormal RBM20 phosphorylation by SRPK3 in the heart would be overcome by ubiquitously expressed kinases, as shown recently for cdc2-like kinases (CLKs) and protein kinase B (AKTs)25 and supported by our in vitro phosphorylation assay. Although RBM20 has not yet been associated with skeletal muscle disease, it has been shown to be DE across different skeletal muscles, where it regulates Z-band and M-band TTN splicing48. In addition, TTN RBM20-mediated splicing regulation is not only skeletal muscle-type specific but also affected by hormone levels49 and circadian rhythm50. Overall, this highlights that RBM20-related pathology is complex and might be caused by the aberrant splicing of target genes through different tissue-specific genomic and nongenomic signaling pathways.
It is not clear whether the SRPK3-related myopathy is caused by the same pathomechanism in mice and humans. This type of discrepancy is not unique, and mouse models do not always recapitulate the human pathology, as seen, for example, in dystroglycanopathies51,52. Nevertheless, it is still possible that the identified Ttn variants might have an effect on the Srpk3 KO mouse line. Interestingly, Srpk3 overexpression in mice results in cardiomyopathy16, not seen in the KO model. Like RBM20, SRPKs exhibit strong spatiotemporal expression profiles13,14,15,16, and their subcellular localization is regulated by their own phosphorylation53 and acetylation54, suggesting that these kinases are involved in tightly controlled and fine-tuned pathways at different developmental stages and in response to external signals17,55. While their role in mRNA regulation is well studied13, it has recently been shown that SRPKs are also involved in ubiquitin signaling56.
We propose that the digenic inheritance of genes involved in post-translational processing and their direct or indirect targets57 may be a model for conditions thus far thought to be monogenic58. Similar digenic inheritance models might also explain incomplete penetrance, such as recently described in spinocerebellar ataxia type 17 (ref. 59). In fact, SRPK3 has previously been suggested as a causative gene in patients with X-linked spinocerebellar ataxia60 and intellectual disability34,61. Notably, it has been proposed that abnormal SRPK-mediated phosphorylation of an E3 ubiquitin ligase might disrupt neurodevelopmental regulation45,56. In addition, haploinsufficiency of the splicing factor SRSF1, a well-known SRPK3 target16, has been newly shown to cause a developmental disorder with intellectual disability62. Based on the findings presented here, it is conceivable that defective or absent phosphorylation activity of this kinase, in combination with a second deficient downstream target gene, could result in these, as well as other, disease phenotypes.
Methods
No statistical methods were used to predetermine the sample size. The experiments were not randomized, and the investigators were not blinded to allocation during experiments and outcome assessment.
Ethics statements
All clinical information and biological material used in this collaborative study were collected after obtaining written informed consent from the patients or their legal guardians. Each sequencing study was approved by the relevant health research authorities (Supplementary Note). Zebrafish were maintained in accordance with UK Home Office regulations, UK Animals (Scientific Procedures) Act 1986, under project licenses 70/7606 and P597E5E82. All animal work was reviewed by The Wellcome Trust Sanger Institute Ethical Review Committee.
Genetic analysis
Genomic DNA from affected individuals was subjected to NGS and analyzed by applying standard filtering criteria (Supplementary Note). SRPK3 and TTN variants were confirmed in the probands and assessed in available family members using Sanger sequencing. Deleteriousness of missense variants was predicted by Combined Annotation Dependent Depletion (CADD, v1.6) scores (https://cadd.gs.washington.edu/)64. NGS data were, when possible, analyzed for CNVs in the TTN gene and its triplicated region visually inspected. Variants in SRPK3 were annotated based on the coding DNA reference sequence NM_014370 and transcript ENST00000370101. Variants in TTN were annotated based on NG 011618.3 or LRG 391 and inferred-complete variant-IC (NM 001267550.1 or ENST00000589042.5), usually referred to as the titin meta-transcript.
Three-dimensional modeling of SRPK3 variants
For the structure-based analysis of SRPK3 variants, a homology model was built using YASARA65 (v15.4.10) with an SRPK1 structural template (5MYV, chain C).
SRPK3 and TTN gene expression and allelic balance analysis
RNA sequencing from muscle biopsies was performed at the Broad Institute Genomics Platform via the Tru-Seq Strand-Specific Large Insert RNA Sequencing protocol, at high coverage (50 M pairs). This included plating, poly-A selection and strand-specific cDNA synthesis, library preparation (450–550 bp insert size) and sequencing (101 bp paired reads). STAR-aligned BAMs were analyzed for gene expression and allelic balance. For gene expression analysis, the featureCounts66 utility from the Subread package (v2.0.0) was used to count reads mapping to annotated genes across the human genome. Resultant counts were processed with edgeR67 (v3.28.1), undergoing normalization with the calcNormFactors function followed by testing for differential gene expression between patient and control samples using the (two-sided) exactTest function68. Results for SRPK3 and TTN were examined, and CPM values, normalized by gene length, were obtained with the cpm function and plotted.
Allelic balance for regions of TTN with evidence of heterozygosity was assessed using the AllelicImbalance69 (v1.24.0) software package, which counts reads at every heterozygous position and applies a chi-square test to determine the statistical significance of any deviation from the expected ratio of reads at each position.
Muscle biopsy and MRI analysis
Muscle biopsies were analyzed following standard histological techniques for light and electron microscopy as part of the diagnostic workup of patients in accredited pathology laboratories. Muscle MRIs were obtained on standard diagnostic scanners using axial T1-weighted scans.
Titin immunoanalysis
Western blotting of titin C-terminal fragments was carried out as previously described20, using two different antibodies raised against the C-terminal M10 Ig domain of titin, rabbit polyclonal M10-1 (ref. 9; 1:300) and mouse monoclonal 11-4-3 (ref. 20; 1:150). For high molecular weight titin western blotting, antibody Z1Z2 (1:1,500, N-terminal, TTN-1; Myomedix) and F146.9B9 (1:1,000, distal I-band titin; Enzo Life Sciences) were used. Snap-frozen muscle biopsies were homogenized in a sample buffer containing 8 M urea, 2 M thiourea, 10% SDS, 0.05 M Tris base and 10% glycerol supplemented with 10% β-mercaptoethanol and heated at 60 °C for 15 min. The soluble fraction was recovered after centrifugation. Equal amounts of muscle protein were loaded into vertical 1% agarose gels, and the run was performed at +8 °C, using 12.5 mA per gel for 6–7 h. The proteins were detected in-gel using SimplyBlue SafeStain (Invitrogen) or blotted to PVDF membranes using CAPS buffer in a Trans-Blot Turbo System with 40 V for 5 h.
Statistical analysis
Three cohorts of patients with genetically confirmed forms of limb-girdle muscular dystrophy, namely LGMD-R1 (n = 170), LGMD-R2 (n = 94) and LGMD-R12 (n = 56) caused by recessive mutations in the CAPN3, DYSF and ANO5 genes, respectively, were identified through the MYO–SEQ Project70. The number of TTNtvs (that is, stop gain, splice sites and frameshift) was counted in each disease control population. A Fisher’s test (two-sided, with no adjustment for multiple comparisons), following the proposal of Agresti and Coull71 to add two successes and two failures to each data set was calculated.
Similar statistical analysis was implemented for the comparison between the number of TTNtvs found in affected and unaffected males carrying a truncating SRPK3 variant (referred to as ‘SRPK3-null males’; Supplementary Note). For the statistical modeling, the MLINK (v5.10) program from the LINKAGE72 package and PSEUDOMARKER73,74 (v2.0) were used. For details, see Supplementary Note.
Zebrafish husbandry and genotyping
Zebrafish were maintained in accordance with UK Home Office regulations, UK Animals (Scientific Procedures) Act 1986, under project licenses 70/7606 and P597E5E82. All animal work was reviewed by The Wellcome Trust Sanger Institute Ethical Review Committee. Zebrafish were maintained at 23.5 °C on a 14 h light/10 h dark cycle. The mutant lines srpk3sa18907 and ttn.1sa5562 were generated by the Zebrafish Mutation Project75. The allele srpk3sa18907 is an essential splice site mutation affecting the donor site of exon 15, and ttn.1sa5562 is a premature stop codon (zebrafish assembly GRCz11 chr9:42861631T>G) in exon 19 of 214, corresponding to amino acid p.Leu1570. Genotyping was carried out as previously described76. Filamentous actin and α-actinin were stained in genotyped larvae at 5 dpf using Alexa Fluor 488 phalloidin (Invitrogen, A12379; 1:80) and mouse monoclonal anti-α-actinin (Sigma, A7811; 1:200).
Zebrafish transcriptomics
In total, 5-dpf larvae were collected as six pools of three embryos per genotype to minimize any differences due to biological variance. A previously described protocol for single embryo RNA extraction77 was optimized for use with pools of zebrafish larvae. Samples were lysed in 110 μl RLT buffer (RNeasy kit, Qiagen) containing 1.1 μl of 14.3 M β-mercaptoethanol (Sigma). The lysate was allowed to bind to 450 μl of Agencourt AMPure XP beads (Beckman Coulter) for 15 min. The tubes were left on a magnet (Invitrogen) until the solutions cleared, and the supernatant was then removed without disturbing the beads. While still on the magnet, the beads were washed three times with 70% ethanol and allowed to dry for 20 min. Total nucleic acid was eluted from the beads following the manufacturer’s instructions and treated with DNase I (New England Biolabs, M0303L). RNA was quantified using a NanoDrop (Thermo Fisher Scientific NanoDrop One Microvolume UV–Vis Spectrophotometer), RNA integrity numbers were checked using a Bioanalyzer (2100 Bioanalyzer System) and sequencing libraries were made using the NEBNext Ultra II DNA Library Prep Kit for Illumina.
Libraries were pooled and sequenced on one lane of NovaSeq 6000 SP PE150 (between 14 million and 29 million reads per sample). RNA-seq data have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under accession E-MTAB-12934. Sequencing data were assessed using FastQC (v0.11.9; https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and aligned to the GRCz11 reference genome using STAR78 (v2.7.3a). Read counts per gene were generated by STAR and used as input for differential expression analysis using the R package DESeq2 (v1.28.1)79. The following model was used for DESeq2: ~Genotype, modeling counts as a function of the genotype. Genes with an adjusted P value of <0.05 using a two-sided Wald test with Benjamini–Hochberg adjustment63 for multiple testing were considered to be DE. Gene sets were analyzed using topGO80 (v2.38.1), and the resulting GO enrichments were visualized using the ClueGO (v2.5.9) plugin for Cytoscape81 (v3.9.1). For analysis of gene expression changes and visualization of data, R (v4.2.0; R Core Team; https://www.r-project.org/) was used, together with the tidyverse82 package (v1.3.1). Differential alternative splicing events were analyzed using rMATS83 (v4.1.2).
Myofiber immunofluorescence
Immunofluorescence staining of zebrafish skeletal myofibers was performed by adapting a protocol previously described84 (Supplementary Note). Myofibers were incubated in primary antibody overnight at 4 °C, washed in 1x Phosphate-Buffered Saline with 0.1% Tween 20 (PBST), then incubated with goat anti-mouse Alexa Fluor 488 secondary antibody (Thermo Fisher Scientific, A11001; 1:500) and goat anti-rabbit Alexa Fluor 594 secondary antibodies (Thermo Fisher Scientific, A11037; 1:250) for 1 h at room temperature. Further washing in PBST was performed before mounting with Vectashield Mounting Medium (Vector Laboratories). Primary antibodies used were rabbit polyclonal anti-sarcomeric α-actinin (Cell Signaling Technologies, 3134; 1:25) and mouse monoclonal anti-titin (Merck, T9030; 1:200). Digital images were captured with Newcastle University Bioimaging Unit’s Nikon A1R point scanning confocal microscope (Nikon) using Nikon Elements AR (v5.21.03).
Transmission electron microscopy (TEM)
Genotyped zebrafish embryos (5 dpf) were fixed with glutaraldehyde (2%) fixative in 0.1 M cacodylate buffer, pH 7.4 (2BScientific, 30450003-1) at 4 °C. Processing for TEM was performed by Newcastle University Electron Microscopy Research Services. Ultrathin sections were stained with heavy metal salts (uranyl acetate and lead citrate). Sections were imaged on a Hitachi HT7800 120 kV TEM using an EMSIS CMOS Xarosa high-resolution camera (Hitachi) and Radius software (v2.0).
Mouse whole-genome sequencing (WGS) analysis
DNA from Srpk3-null (KO)16 and WT (129s6/SvEvTAC background) mice were subjected to WGS at deCODE Genetics. Fastq files were mapped using the BBmap suite (v38.69) against the mouse Ttn gene sequence (USCS, GRCm38/mm10). Variants (with a read frequency of >20%) were called using Varscan 2 (v2.3.7) for each sample, and BCFTools (v1.9) was used to merge all samples by group (WT and KO). The output variants present in the WT and KO groups were annotated using the Variant Effect Predictor tool (Ensembl, https://www.ensembl.org/info/docs/tools/vep/index.html, https://www.ensembl.org/Tools/VEP). Variants were compared to the reference 129s6/SvEvTAC background.
Constructs
To create the RBM20517–664-V5 reporter, the cDNA fragment coding amino acids 517–664 of mouse RBM20 was cloned into pcDNA3.1D/V5-His-TOPO in frame with C-terminal V5 and His6 tags using the pcDNA3.1 Directional TOPO Expression Kit (Thermo Fisher Scientific, K490001). The pcDNA5-FRT/TO-GFP-SRPK3 construct (DU 25699), expressing N-terminally GFP human SRPK3, was obtained from the Medical Research Council Protein Phosphorylation and Ubiquitylation Unit Reagents and Services (University of Dundee, Scotland).
Phosphorylation assay
The 293T cells were transfected with the RBM20517–664-V5 reporter construct together with either GFP-SRPK3 or an empty vector (pcDNA5/TO), collected after 2 d of expression and frozen at −80 °C. The cells were lysed for 15 min on ice in 1× NEBuffer Pack for Protein Metallophosphatases (50 mM HEPES (pH 7.5), 100 mM NaCl, 2 mM DTT, 0.01% Brij 35; New England Biolabs) supplemented with 1 mM MnCl2, 1% Triton X-100, 1× Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific), 4 mM β-glycerophosphate, 10 mM sodium pyrophosphate, 4 mM sodium orthovanadate and 2 mM sodium fluoride. Insoluble material was pelleted at 4 °C for 15 min at 15,700g. The supernatants were divided into three reactions (U, untreated; N, no phosphatase; P, phosphatase) and combined 1:1 with dephosphorylation buffer (1× NEBuffer pack for protein metallophosphatases, 1 mM MnCl2) alone (U, N) or with NEB lambda protein phosphatase (P; final concentration 6,667 units ml−1). The U reactions were immediately mixed with 2× SDS sample buffer and heated for 5 min at 95 °C, whereas the N and P reactions were incubated for 30 min at 30 °C before SDS sample preparation. The samples were run in standard SDS–PAGE in 4–20% TGX minigels (Bio-Rad), transferred on nitrocellulose membranes and stained with antibodies against the V5 tag (Thermo Fisher Scientific, R960-25, SV5-Pk1; 1:5,000) and GFP (Thermo Fisher Scientific, A-11122; 1:5,000).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Due to privacy, ethical and legal issues de-identified patient genomic, transcriptomic and phenotypic data that supports the findings of this study can only be available from the corresponding author upon reasonable request. Zebrafish RNA-seq data can be accessed in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under accession E-MTAB-12934. Mouse WGS data and human RNA-seq data can be accessed in the Sequence Read Archive under accession (PRJNA1027609 and PRJNA1027754, respectively). Control frequencies and variant information were extracted from gnomAD (v2.1.1; https://gnomad.broadinstitute.org). TTN variant information was obtained from the Leiden Open Variation Database (https://databases.lovd.nl/shared/genes/TTN). Source data are provided with this paper.
Code availability
All software used to analyze the study data are listed in the Methods and in the Nature Research Reporting Summary and are publicly available.
References
Waldrop, M. A. et al. Diagnostic utility of whole exome sequencing in the neuromuscular clinic. Neuropediatrics 50, 96–102 (2019).
Deltas, C. Digenic inheritance and genetic modifiers. Clin. Genet. 93, 429–438 (2018).
Van der Ven, P. F., Bartsch, J. W., Gautel, M., Jockusch, H. & Fürst, D. O. A functional knock-out of titin results in defective myofibril assembly. J. Cell Sci. 113, 1405–1414 (2000).
Roberts, A. M. et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 7, 270ra6 (2015).
Savarese, M. et al. The complexity of titin splicing pattern in human adult skeletal muscles. Skelet. Muscle 8, 11 (2018).
Savarese, M. et al. Genotype-phenotype correlations in recessive titinopathies. Genet. Med. 22, 2029–2040 (2020).
Evilä, A. et al. Targeted next-generation sequencing reveals novel TTN mutations causing recessive distal titinopathy. Mol. Neurobiol. 54, 7212–7223 (2017).
Oates, E. C. et al. Congenital titinopathy: comprehensive characterization and pathogenic insights. Ann. Neurol. 83, 1105–1124 (2018).
Hackman, P. et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am. J. Hum. Genet. 71, 492–500 (2002).
Pfeffer, G. et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain 135, 1695–1713 (2012).
Herman, D. S. et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 366, 619–628 (2012).
Savarese, M. et al. Interpreting genetic variants in titin in patients with muscle disorders. JAMA Neurol. 75, 557–565 (2018).
Giannakouros, T., Nikolakaki, E., Mylonis, I. & Georgatsou, E. Serine-arginine protein kinases: a small protein kinase family with a large cellular presence. FEBS J. 278, 570–586 (2011).
Gui, J. F., Lane, W. S. & Fu, X. D. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature 369, 678–682 (1994).
Wang, H. Y. et al. SRPK2: a differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J. Cell Biol. 140, 737–750 (1998).
Nakagawa, O. et al. Centronuclear myopathy in mice lacking a novel muscle-specific protein kinase transcriptionally regulated by MEF2. Genes Dev. 19, 2066–2077 (2005).
Fu, X. D. & Ares, M. Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 15, 689–701 (2014).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Norton, N. et al. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ. Cardiovasc. Genet. 6, 144–153 (2013).
Evilä, A. et al. Atypical phenotypes in titinopathies explained by second titin mutations. Ann. Neurol. 75, 230–240 (2014).
Akinrinade, O., Koskenvuo, J. W. & Alastalo, T. P. Prevalence of titin truncating variants in general population. PLoS ONE 10, e0145284 (2015).
Shih, Y. H. et al. Exon- and contraction-dependent functions of titin in sarcomere assembly. Development 143, 4713–4722 (2016).
Li, S., Guo, W., Dewey, C. N. & Greaser, M. L. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 41, 2659–2672 (2013).
Murayama, R. et al. Phosphorylation of the RSRSP stretch is critical for splicing regulation by RNA-binding motif protein 20 (RBM20) through nuclear localization. Sci. Rep. 8, 8970 (2018).
Sun, M. et al. SR protein kinases regulate the splicing of cardiomyopathy-relevant genes via phosphorylation of the RSRSP Stretch in RBM20. Genes (Basel) 13, 1526 (2022).
Castiglione, A. & Moller, C. Usher syndrome. Audiol. Res. 12, 42–65 (2022).
Liu, X. Z. et al. Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum. Genet. 125, 53–62 (2009).
Yang, Z. et al. Digenic heterozygous mutations of KCNH2 and SCN5A induced young and early-onset long QT syndrome and sinoatrial node dysfunction. Ann. Noninvasive Electrocardiol. 27, e12889 (2022).
Chen, Q. et al. Digenic variants in the TTN and TRAPPC11 genes co-segregating with a limb-girdle muscular dystrophy in a han Chinese family. Front. Neurosci. 15, 601757 (2021).
Peddareddygari, L. R., Oberoi, K. & Grewal, R. P. Limb girdle muscular dystrophy due to digenic inheritance of DES and CAPN3 mutations. Case Rep. Neurol. 10, 272–278 (2018).
Lee, Y. et al. TIA1 variant drives myodegeneration in multisystem proteinopathy with SQSTM1 mutations. J. Clin. Invest. 128, 1164–1177 (2018).
Lemmers, R. J. et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 44, 1370–1374 (2012).
Zhang, Y. et al. RBM20 phosphorylation and its role in nucleocytoplasmic transport and cardiac pathogenesis. FASEB J. 36, e22302 (2022).
Kim, C. H. et al. Eye movement defects in KO zebrafish reveals SRPK3 as a causative gene for an X-linked intellectual disability. Preprint at Research Square https://doi.org/10.21203/rs.3.rs-2683050/v1 (2023).
Rees, M. et al. Making sense of missense variants in TTN-related congenital myopathies. Acta Neuropathol. 141, 431–453 (2021).
Dabby, R. et al. Adult onset limb-girdle muscular dystrophy—a recessive titinopathy masquerading as myositis. J. Neurol. Sci. 351, 120–123 (2015).
Zheng, W. et al. Identification of a novel mutation in the titin gene in a Chinese family with limb-girdle muscular dystrophy 2J. Mol. Neurobiol. 53, 5097–5102 (2016).
Nallamilli, B. R. et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann. Clin. Transl. Neurol. 5, 1574–1587 (2018).
Rich, K. A. et al. Novel heterozygous truncating titin variants affecting the A-band are associated with cardiomyopathy and myopathy/muscular dystrophy. Mol. Genet. Genom. Med. 8, e1460 (2020).
Guo, W. et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 18, 766–773 (2012).
Koelemen, J., Gotthardt, M., Steinmetz, L. M. & Meder, B. RBM20-related cardiomyopathy: current understanding and future options. J. Clin. Med. 10, 4101 (2021).
Brauch, K. M. et al. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J. Am. Coll. Cardiol. 54, 930–941 (2009).
Zhu, C., Yin, Z., Tan, B. & Guo, W. Insulin regulates titin pre-mRNA splicing through the PI3K-Akt-mTOR kinase axis in a RBM20-dependent manner. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 2363–2371 (2017).
Fenix, A. M. et al. Gain-of-function cardiomyopathic mutations in RBM20 rewire splicing regulation and re-distribute ribonucleoprotein granules within processing bodies. Nat. Commun. 12, 6324 (2021).
Beqqali, A. et al. A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc. Res. 112, 452–463 (2016).
Gaertner, A. et al. The combined human genotype of truncating TTN and RBM20 mutations is associated with severe and early onset of dilated cardiomyopathy. Genes (Basel) 12, 883 (2021).
Van den Hoogenhof, M. M. G. et al. RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 138, 1330–1342 (2018).
Chen, Z. et al. Z-band and M-band titin splicing and regulation by RNA binding motif 20 in striated muscles. J. Cell. Biochem. 119, 9986–9996 (2018).
Maimaiti, R., Zhu, C., Zhang, Y., Ding, Q. & Guo, W. RBM20-mediated pre-mRNA splicing has muscle-specificity and differential hormonal responses between muscles and in muscle cell cultures. Int. J. Mol. Sci. 22, 2928 (2021).
Riley, L. A. et al. The skeletal muscle circadian clock regulates titin splicing through RBM20. eLife 11, e76478 (2022).
White, J. K. et al. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell 154, 452–464 (2013).
Brown, S. C., Fernandez-Fuente, M., Muntoni, F. & Vissing, J. Phenotypic spectrum of α-dystroglycanopathies associated with the c.919T>a variant in the FKRP gene in humans and mice. J. Neuropathol. Exp. Neurol. 79, 1257–1264 (2020).
Zhou, Z. et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol. Cell 47, 422–433 (2012).
Wang, C. et al. SRPK1 acetylation modulates alternative splicing to regulate cisplatin resistance in breast cancer cells. Commun. Biol. 3, 268 (2020).
Nikolakaki, E., Sigala, I. & Giannakouros, T. Good cop, bad cop: the different roles of SRPKs. Front. Genet. 13, 902718 (2022).
Bustos, F. et al. Functional diversification of SRSF protein kinase to control ubiquitin-dependent neurodevelopmental signaling. Dev. Cell 55, 629–647 (2020).
Navarro, C. L. et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum. Mol. Genet. 13, 2493–2503 (2004).
Zhang, H. et al. ENAM mutations and digenic inheritance. Mol. Genet. Genomic Med. 7, e00928 (2019).
Barbier, M. et al. Intermediate repeat expansions of TBP and STUB1: genetic modifier or pure digenic inheritance in spinocerebellar ataxias? Genet. Med. 25, 100327 (2023).
Thomas, M. G. et al. Whole exome sequencing identifies a new splicing factor gene causative of X-linked spinocerebellar ataxia. Invest. Ophthalmol. Vis. Sci. 56, 3994 (2015).
Niranjan, T. S. et al. Affected kindred analysis of human X chromosome exomes to identify novel X-linked intellectual disability genes. PLoS ONE 10, e0116454 (2015).
Bogaert, E. et al. SRSF1 haploinsufficiency is responsible for a syndromic developmental disorder associated with intellectual disability. Am. J. Hum. Genet. 110, 790–808 (2023).
Hochberg, Y. & Benjamini, Y. More powerful procedures for multiple significance testing. Stat. Med. 9, 811–818 (1990).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Krieger, E. et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins 77, 114–122 (2009).
Liao, Y., Smyth, G. K. & Shi, W. The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Robinson, M. D. & Smyth, G. K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9, 321–332 (2008).
Gadin, J. R., van’t Hooft, F. M., Eriksson, P. & Folkersen, L. AllelicImbalance: an R/bioconductor package for detecting, managing, and visualizing allele expression imbalance data from RNA sequencing. BMC Bioinformatics 16, 194 (2015).
Töpf, A. et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness. Genet. Med. 22, 1478–1488 (2020).
Agresti, A. & Coull, B. A. Order-restricted tests for stratified comparisons of binomial proportions. Biometrics 52, 1103–1111 (1996).
Lathrop, G. M., Lalouel, J. M., Julier, C. & Ott, J. Strategies for multilocus linkage analysis in humans. Proc. Natl Acad. Sci. USA 81, 3443–3446 (1984).
Gertz, E. M. et al. PSEUDOMARKER 2.0: efficient computation of likelihoods using NOMAD. BMC Bioinformatics 15, 47 (2014).
Hiekkalinna, T. et al. PSEUDOMARKER: a powerful program for joint linkage and/or linkage disequilibrium analysis on mixtures of singletons and related individuals. Hum. Hered. 71, 256–266 (2011).
Kettleborough, R. N. et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature 496, 494–497 (2013).
Dooley, C. M. et al. Multi-allelic phenotyping—a systematic approach for the simultaneous analysis of multiple induced mutations. Methods 62, 197–206 (2013).
Wali, N., Merteroglu, M., White, R. J. & Busch-Nentwich, E. M. Total nucleic acid extraction from single zebrafish embryos for genotyping and RNA-seq. Bio Protoc. 12, e4284 (2022).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Alexa, A., Rahnenfuhrer, J. & Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607 (2006).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Whickham, H., Averick, M., Bryan, J. & Chang, W. Welcome to Tidyverse. J. Open Source Softw. 4, 1986 (2019).
Shen, S. et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-seq data. Proc. Natl Acad. Sci. USA 111, E5593–E5601 (2014).
Horstick, E. J., Gibbs, E. M., Li, X., Davidson, A. E. & Dowling, J. J. Analysis of embryonic and larval zebrafish skeletal myofibers from dissociated preparations. J. Vis. Exp. 13, e50259 (2013).
Acknowledgements
We acknowledge H. Luque, L. Phillips, J. Casement, O. Magnuson, D. Nguyen and Y. Hu for technical support; R. García-Tercero and C. Díaz for sample collection; E. Zorio, M.E. Leach, D. Bharucha-Goebel, J. Dastgir and C. Konersman for clinical expertise and M. Gautel for helpful advice. We also thank CureCMD for their help in patient recruitment and the patients for donating their samples.
The research leading to these results has received funding from the European Community’s Seventh Framework Program (FP7/2007-2013; 2012-305121) ‘Integrated European—omics research project for diagnosis and therapy in rare neuromuscular and neurodegenerative diseases (NEUROMICS)’ (to A. Töpf, V.S., I.T.Z. and F.M.); the European Union’s Horizon 2020 research and innovation program (Solve-RD project; 779257 to A. Töpf); Muscular Dystrophy UK and Muscular Dystrophy Association US (mda577346 to F.M.); Päulon Säätiö (to M. Savarese); Academy of Finland, Sigrid Juselius Foundation (to B.U.); core funding to the Sanger Institute by the Wellcome Trust (098051 and 206194 to E.M.B.-N., J.P. and N.W.); EURO-NMD and Fundación Gemio (to J.J.V., N.M. and P.M.); Intramural Research Grant (2-5, 29-4) for Neurological and Psychiatric Disorders of NCNP and AMED (JP20ek0109490h0001 to I.N.); Inserm, CNRS, University of Strasbourg, Labex INRT (ANR-10-LABX-0030 and ANR-10-IDEX-0002-02), France Génomique (ANR-10-INBS-09) and Fondation Maladies Rares for the ‘Myocapture’ sequencing project, AFM-Téléthon (22734), the European Joint program (EJPRD2019-126 IDOLS-G and ANR-19-RAR4-0002 to J.L., X.L. and V.B.); Intramural funds from the NIH National Institute of Neurological Disorders and Stroke (to C.G.B.); the Dutch Princess Beatrix Muscle Fund and the Dutch Spieren voor Spieren Muscle fund (to C.E.E.); PI16/00316 supported by the Instituto de Salud Carlos III (ISCIII), Madrid and the Generalitat Valenciana (grant PROMETEO/2019/075 to N.M.); Australian NHMRC Neil Hamilton Fairley Early Career Research Fellowship (GNT1090428 to E.C.O.); Starship Foundation A+7340 (to G.L.O.); Early Career Award from the Thrasher Research Fund (to S.S.); U54 HD090255 from the NIH Eunice Kennedy Shriver National Institute of Child Health and Human Development (to A.H.B.); Wellcome Center for Mitochondrial Research (203105/Z/16/Z), the Mitochondrial Disease Patient Cohort (UK; G0800674), the Medical Research Council International Center for Genomic Medicine in Neuromuscular Disease (MR/S005021/1), the Medical Research Council (MR/W019027/1), the Lily Foundation, Mito Foundation, the Pathological Society, the UK NIHR Biomedical Research Center for Ageing and Age-related Disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust and the UK NHS Highly Specialized Service for Rare Mitochondrial Disorders of Adults and Children (to R.W.T.). MYO–SEQ was funded by Sanofi Genzyme, Ultragenyx, LGMD2I Research Fund, Samantha J Brazzo Foundation, LGMD2D Foundation, Kurt+Peter Foundation, Muscular Dystrophy UK and Coalition to Cure Calpain 3. Sequencing and analysis for relevant families (Supplementary Note) were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and were funded by the National Human Genome Research Institute, the National Eye Institute and the National Heart, Lung and Blood Institute under grant UM1 HG008900 and the National Human Genome Research Institute under grants U01HG0011755 and R01 HG009141. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. DNA samples for NeurOmics and MYO–SEQ were provided by the John Walton Muscular Dystrophy Research Center Biobank. This facility is supported by the NIHR Newcastle Biomedical Research Center. Newcastle University’s Electron Microscopy Research Services and equipment Hitachi HT7800 120 kV TEM microscope are funded by BBSRC grant reference BB/R013942/1.
Author information
Authors and Affiliations
Contributions
A. Töpf, B.U., E.M.B.-N., F.M., H.J.C., J.L., I.N. and V.S. designed the study. Data were collected by A. Töpf, D.C., A.H.B., A.C., A.M., A.N., A.O’D.-L., A. Sarkozy, A. Tuite, B.A.P., B.V., C.E.E., C.A.G., C.G., C.G.B., C.M.-B., C.V., D.G.M., E.C.O., E.H., E.-J.K., E.M., E.M.E., E.O’H., E.W., G.L.O’G., G.T., G.V., H.J., H.J.C., I.B., I.N., I.S., I.T.Z., J.D., J.D.-M., J.J.V., J.L., J.P., J.S., J.V.W., K.G.C., K.Y., M.H., M.M., M.R.D., M.R.F., M. Savarese, M. Schouten, M.-T.C., N.C.V., N.M., N.O., N.B.R., N.W., O.N., P.M., P.V.d.B., R.B., R.P., R.W.T., S.A.-H., S.T.C., S.D., S.L.S., S.P., S.S., T.E.M., V.B., V.D.L., W.N.L., X.L. and Y.S. Formal analysis was carried out by A. Töpf, A. Smolnikov, B.B.C., B.V., D.C., E.M.B.-N., H.J.C., I.M.S., J.P., K.M.L., M.M., N.W., P.H.J., R.J.W. and S.L.S. Visualization of data was done by A. Töpf, A. Smolnikov, A.V., D.C., I.M.S., I.T.Z., J.D.M., J.S., J.V.W., M.M., P.H.J., R.J.W. and S.B. A. Töpf wrote the original draft. All authors reviewed and edited the draft and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks James Dowling and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. This article has been peer-reviewed as part of Springer Nature’s Guided Open Access initiative. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 RNA analysis of SRPK3 truncating variants.
a, RT-PCR of muscle-derived cDNA from patient CII:2 carrying a donor splice site variant (c.1144+1G>A). PCR amplification with Ex4F/Ex11R primers showed that the variant leads to the exclusion of exon 10 and results in a frameshift change (p.Asp284_Thr383delinsAla). b, RT-PCR of muscle-derived cDNA from patient SII:1 carrying an extended splice site variant (c.774+5G>C); top: using primers in exons flanking the variant (Ex6F 5′-CGTGAAGAGCATCGTGAGG and Ex10R 5′- GCCCCCGTCTAGTCTCAAG) a single band was detected in the patient (P), corresponding to abnormal exon 8 skipping (r.749_774del, p.Val250Alafs*4); bottom: using the same forward primer in exon 6 and a reverse primer in intron 8 (In8R 5′- GACGGCCCGGTACTGCCGAGTCTG), two and three bands were detected in the proband and control samples, respectively. The larger bands correspond to gDNA; the band at 373 bp corresponds to abnormal retention of both intron 7 and intron 8 in the proband (r.748_749ins[748+1_749-1]; r.774_775ins[g>c;774+1_775-1], p.Val250Glyfs*46). The lower band corresponds to a natural missplicing event leading to retention of intron 8 observed both in the patient and controls (r.774_775ins[g>c;774+1_775-1], p.Val250Glyfs*46). c, Sashimi plot for RNA sequencing data from muscle tissue from patient LII:1 carrying a donor splice variant (c.190+2T>C). The plot shows skipping of exon 2, leading to an out-of-frame mRNA. Any SPRK3 transcripts escaping nonsense-mediated decay will encode a truncated protein lacking the kinase domain. RT-PCR amplification was performed at least in duplicate.
Extended Data Fig. 2 SRPK3 mRNA expression levels in patients with SRPK3 truncating variants.
RNA sequencing data from patients carrying SRPK3 truncating variants (LII:1, DII:1 and YII:3) were analyzed for SRPK3 expression between patient (n = 3) and control (n = 6) samples. Counts per million (CPM) values were normalized by gene length. The boxes represent the first and third quartiles (25% and 75% percentile) with the center line at the median value. The whiskers extend from the hinge to the furthest value not beyond 1.5 times the interquartile range from the hinge. Differential expression was performed in edgeR using a two-sided exact test, with no adjustment for multiple comparisons. Uncorrected P-value = 7.106 × 10−11.
Extended Data Fig. 3 Localization and 3D modeling of SRPK3 variants.
a, Localization of SRPK3 variants. Kinase domains are indicated in green. Missense changes are shown in green, truncating variants (nonsense, splice sites and frameshift) in black and the in-frame variant in red. Two splice site variants were identified at position c.749-2 (shown as X252_splice). b, For the structure-based analysis of SRPK3 variants, a homology model was built using YASARA (v15.4.10) with a SRPK1 structural template (5MYV, chain C). Amino acid position for changes p.Arg131Pro, p.Pro135His, p.Gly157Arg, p.Leu196Pro, p.Arg430Gln, p.Val434Glu, p.Asp445Asn, p.Glu455Lys, p.Arg553Trp and the p.Lys405_Ile406del are indicated. p.Arg131Pro + p.Pro135His: Pro135 is located in a loop where it bends the loop in a way to allow for stabilizing interactions, where a change to His will lead to disruption of local loop orientation. Arg131 is located at the C-terminal of the alpha-helix, close to Pro135. In an additive fashion, the introduction of Arg131Pro will probably destabilize local structure even further. p.Gly157Arg: will force changes in backbone orientation for residues in the loop and surrounding sheet structures, leading to local stability issues by forcing surrounding residues to adopt orientations that impair favorable interactions. p.Leu196Pro: will lead to a disruption of the helical structure and decrease protein stability. p.Arg430Gln: Arg430 plays a stabilizing role by interacting with the negatively charged residues Asp474 and Asp423. Loss of this positive charge will lead to destabilizing effects. p.Val434Glu: introduces a negatively charged residue that will likely disrupt the wild-type charged interaction network of Arg430, Asp474 and Asp423. p.Asp445Asn: loss of negative charge, loss of hydrogen bond with backbone Thr211, leading to a destabilizing effect. p.Glu455Lys: change of a highly conserved negative charge into positive charge. Forms a hydrogen bond with Tyr429, which is lost when mutated to Lys. Probably destabilizing, although the role of the negative charge is not clear. p.Arg553Trp: Arg533 forms a salt bridge with Glu433, stabilizing local structure. Losing that salt bridge will destabilize. Additionally, exchanging a large, positively charged residue for a bulky very hydrophobic residue will lead to additional destabilization. p.Lys405_Ile406del (also annotated as p.Lys402Ile403del): results in a deletion of two residues (Lys-Ile) in a short repeat sequence (Lys-Ile-Lys-Ile-Lys-Ile). Protein structure modeling suggests that the deleted residues are Lys402 and Ile403. The modeled structure shows Asp401 reoriented into a position originally occupied by Ile403. This will destabilize local structure. Additionally, it results in the loss of the salt bridge between Asp401 and Arg193, which also contributes to a destabilizing effect.
Extended Data Fig. 4 Distribution of TTN variants identified in the SRPK3/TTN cohort.
TTN truncating variants (nonsense, splice sites and frameshift) are shown in black and missense variants in green. No missense variants in TTN exons 344 or 364, associated with HMERF and tibial muscular dystrophy, respectively, were found. TTN variants were located mainly in the A-band and I-band; however, no clustering was observed. Two frameshift variants occurred in meta transcript only exons (in pink).
Extended Data Fig. 5 TTN mRNA expression levels in SRPK3/TTN patients.
RNA sequencing data from SRPK3/TTN myopathy patients (LII:1, DII:1 and YII:3) were analyzed for TTN expression between patient (n = 3) and control (n = 6) samples. Counts per million (CPM) values were normalized by gene length. The boxes represent the first and third quartiles (25% and 75% percentile) with the center line at the median value. The whiskers extend from the hinge to the furthest value not beyond 1.5 times the interquartile range from the hinge. Differential expression was performed in edgeR using a two-sided exact test, with no adjustment for multiple comparisons. Uncorrected P-value = 0.0008015. This was likely not due to nonsense-mediated mRNA decay, as there was no evidence of allele-specific expression at any of the heterozygous TTN loci examined.
Extended Data Fig. 6 TTNtv in other muscle disease populations.
Comparison between the number of TTNtv (stop gain, splice sites and frameshift variants) in the SRPK3/TTN myopathy families (n = 25) and three cohorts of patients with genetically confirmed forms of limb girdle muscular dystrophy: LGMD-R1 (n = 170), LGMD-R2 (n = 94) and LGMD-R12 (n = 56). A Fisher’s test (two-sided, with no adjustment for multiple comparisons), following the proposal of Agresti and Coull to add two successes and two failures to each data set was calculated. P-values: (*) = 6.13 × 10−19, (**) = 9.18 × 10−17 and (***) = 4.89 × 10−12.
Extended Data Fig. 7 Effect of the zebrafish srpk3 sa18907 mutation on mRNA.
a, The mutation lies between exon 15 and 16 in the zebrafish genomic sequence. RT-PCR of muscle-derived cDNA from zebrafish (wild-type, heterozygous and homozygous for the sa18907 mutation) showed three different sized products (primers: Fwd 5′- CTGCTGACATATGGAGCACTG and Rev 5′-GGATACTAAATGTCCCGTAGGTTG). Wild-type samples showed the expected 401-bp product (middle band). The mutation results in aberrant splicing of srpk3 with partial retention of intron 15 (top band) seen both in the homozygous and heterozygous state, or loss of exon 15 (lower band) only seen in the homozygous mutants. Representative of two experiments. b, Sashimi plot for RNA sequencing data from srpk3 mutant and wild-type zebrafish, also showing skipping of exon 15, as well as several forms of partial retention of intron 15.
Extended Data Fig. 8 ttn.1-null zebrafish shows a severe skeletal muscle phenotype.
a–f, Lateral view of Alexa Fluor phalloidin filamentous actin (green) and α-actinin Z-band marker (red) staining in skeletal fast muscle fibers in wild-type (a,d), srpk3+/+;ttn.1−/− (b,e) and srpk3−/−;ttn.1−/− (c,f) larvae at 5 dpf. Compared to wild-type fish (a,d), the muscle fiber structure was largely lost in the ttn.1-null zebrafish, regardless of the srpk3 status (that is both srpk3+/+;ttn.1−/− and srpk3−/−;ttn.1−/−). Scale bars are 25 µm. Representative images from >15 pooled fish per genotype.
Extended Data Fig. 9 mRNA expression in the srpk3 and ttn zebrafish models.
a, Transcriptome data showed that ttn.1 mRNA (ENSDARG00000000563) expression is equally reduced in the heterozygous (srpk3+/+; ttn.1+/−) and the double mutants (srpk3−/−; ttn.1+/−) when compared to the wild-type ttn.1. P-values: (*) sprk3+/+; ttn.1+/− vs. srpk3+/+; ttn.1+/+ = 0.024, (**) sprk3−/−; ttn.1+/− vs. srpk3−/−; ttn.1+/+ = 0.004, (***) sprk3−/−; ttn.1+/− vs. srpk3+/+; ttn.1+/+ = 3.221 × 10−4. b, srpk3 mRNA (ENSDARG00000005916) was upregulated in the srpk3−/− zebrafish mutants (both srpk3−/−; ttn.1+/+ and srpk3−/−; ttn.1+/−), likely as a compensatory effect. P-values: (**) sprk3−/−; ttn.1+/− vs. srpk3+/+; ttn.1+/+ = 0.005, (***) sprk3−/−; ttn.1+/+ vs. srpk3+/+; ttn.1+/+ = 5.178 × 10−4. The boxes represent the first and third quartiles (25% and 75% percentile) with the center line at the median value. The whiskers extend from the hinge to the furthest value not beyond 1.5 times the interquartile range from the hinge. Any outlier values beyond 1.5 times the interquartile range are plotted as individual points. Differential expression was done using a two-sided Wald test with Benjamini-Hochberg adjustment for multiple testing. n = 6 for each condition.
Extended Data Fig. 10 Overlap of differentially expressed (DE) genes annotated to enriched Gene Ontology (GO) terms.
The table shows the top enriched GO terms from the comparisons of srpk3−/−; ttn.1+/− and srpk3−/−; ttn.1+/+ embryos to wild-type (P < 0.001 in one of the comparisons). The topGO P-value columns show the P-value from the enrichment test for each comparison. The dots in the number of DE genes columns represent the number of DE genes annotated to the term that appear either in srpk3−/−; ttn.1+/− alone (blue), srpk3−/−; ttn.1+/+ alone (red) or in both (purple). For most GO terms, most of the DE genes causing the enrichment are shared between both lists. GO term enrichment was done using the topGO package using a one-sided Fisher’s exact test without adjustment for multiple testing.
Supplementary information
Supplementary Information
Supplementary Note.
Supplementary Tables
Supplementary Tables 1–3.
Supplementary Data 1
Zebrafish mRNA profile. DE genes from comparisons of various genotypes as determined by DESeq2 (adjusted P value 0.05) using a two-sided Wald test with Benjamini–Hochberg adjustment for multiple testing. Gene = ensembl gene id; pval = DESeq2 P value; adjp = Benjamini–Hochberg adjusted P values for multiple testing; Chr, Start, End, Strand = genomic location of gene; biotype = ensembl gene biotype; name = gene name; description = gene description from ensembl; the remaining columns represent raw counts and DESeq2 normalized counts for each individual sample.
Supplementary Data 2
Editorial assessment report.
Source data
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 6
Unprocessed western blots.
Source Data Extended Data Fig. 1
Unprocessed gels.
Source Data Extended Data Fig. 7
Unprocessed gels.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Töpf, A., Cox, D., Zaharieva, I.T. et al. Digenic inheritance involving a muscle-specific protein kinase and the giant titin protein causes a skeletal muscle myopathy. Nat Genet 56, 395–407 (2024). https://doi.org/10.1038/s41588-023-01651-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-023-01651-0
