
Abstract
Owing to its high thermal and electrical conductivities, its ductility and its overall non-toxicity1,2,3, copper is widely used in daily applications and in industry, particularly in anti-oxidation technologies. However, many widespread anti-oxidation techniques, such as alloying and electroplating1,2, often degrade some physical properties (for example, thermal and electrical conductivities and colour) and introduce harmful elements such as chromium and nickel. Although efforts have been made to develop surface passivation technologies using organic molecules, inorganic materials or carbon-based materials as oxidation inhibitors4,5,6,7,8,9,10,11,12, their large-scale application has had limited success. We have previously reported the solvothermal synthesis of highly air-stable copper nanosheets using formate as a reducing agent13. Here we report that a solvothermal treatment of copper in the presence of sodium formate leads to crystallographic reconstruction of the copper surface and formation of an ultrathin surface coordination layer. We reveal that the surface modification does not affect the electrical or thermal conductivities of the bulk copper, but introduces high oxidation resistance in air, salt spray and alkaline conditions. We also develop a rapid room-temperature electrochemical synthesis protocol, with the resulting materials demonstrating similarly strong passivation performance. We further improve the oxidation resistance of the copper surfaces by introducing alkanethiol ligands to coordinate with steps or defect sites that are not protected by the passivation layer. We demonstrate that the mild treatment conditions make this technology applicable to the preparation of air-stable copper materials in different forms, including foils, nanowires, nanoparticles and bulk pastes. We expect that the technology developed in this work will help to expand the industrial applications of copper.
Similar content being viewed by others

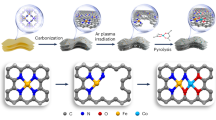
Main
The surface coordination passivation on Cu was first created by hydrothermally treating Cu foils in an aqueous solution of sodium formate at 200 °C. As shown in Fig. 1a, the surface of untreated Cu turned fully dark after being kept in 0.1 M NaOH at 25 °C for 8 h. By contrast, the Cu foil after formate treatment (denoted as Cu-FA) retained its metallic lustre under the same conditions, and outperformed even widely used Cu-alloys (that is, brass and bronze), graphene-coated Cu and benzotriazole (BTA)-treated Cu (Extended Data Fig. 1a, b). As revealed by optical, scanning electron microscopy (SEM), Raman and X-ray diffraction (XRD) analyses (Fig. 1b, c, Extended Data Fig. 1a–d), whereas the surfaces of brass, bronze and graphene-coated Cu foils were heavily oxidized, with the formation of black-coloured CuO after air exposure in NaOH, no obvious oxidation species were detected on Cu-FA.

a, Photographs of formate-modified Cu foil (Cu-FA), Cu foil, brass and graphene-coated Cu foils (Cu-G) before (top) and after (bottom) exposure to 0.1 M NaOH for 8 h. b, Optical microscope photographs and corresponding Raman spectra of Cu-FA (left) and Cu foils (right) taken after NaOH exposure for 8 h. c, Representative SEM images of Cu-FA and Cu foils before (top) and after (bottom) exposure to NaOH for 8 h.
The hydrothermal temperature of the formate treatment is critical for building up an anti-corrosive surface on Cu. Lowering the temperature from 200 °C to 160 °C failed to create effective passivation (Extended Data Fig. 1e). By switching the treatment to solvothermal conditions with the presence of sodium formate and oleylamine in a mixed solvent of dimethylformamide (DMF) and H2O, the passivation on Cu (Extended Data Fig. 1f–j) was readily developed at 160 °C. Quantitative electrochemical measurements (see Extended Data Fig. 1h, Methods) demonstrated that the oxidative corrosion rate of Cu-FA in 0.1 M NaOH was reduced to 3.89 μm yr−1, corresponding to a 20-fold enhancement in anti-corrosion performance compared to bare Cu (78.2 μm yr−1)14,15,16. The anti-corrosion performance achieved by the formate treatment was much better than that reported for small organic molecules4,10,16. The corrosion rate of Cu-FA was one order of magnitude lower than that of Cu foil treated by 1-dodecathiol (Cu-DT; 50.4 μm yr−1) and BTA (Cu-BTA; 59.7 μm yr−1) (Extended Data Fig. 2a). In addition, the passivation layer was effective at preventing the oxidation of Cu under other harsh conditions (Extended Data Fig. 2b–d). After electrochemical tests (1 h in NaOH), whereas the untreated Cu foil was heavily corroded, with the formation of a rough surface containing oxidized products, the surface of Cu-FA remained almost unchanged. Electrochemical measurements revealed a much enhanced corrosion resistance of Cu in 1 M NaOH after the formate treatment. The surface of Cu-FA remained intact after exposure to H2O2 (30%) for 15 min. Oxidation started to take place after 60 min. In comparison, the surface of untreated Cu was severely oxidized after 15 min. After 1 h of heating at 160 °C in air, the surface passivation layer was still highly effective at preventing oxidation of Cu in alkaline conditions. Moreover, as revealed by a home-built integrated scanning reference electrode technique and scanning tunnelling microscopy (SRET/STM) system, even when Cu-FA was scratched, oxidative corrosion was restricted locally to the breached areas and was not accelerated (Extended Data Fig. 2e), indicating that the method developed here is superior to techniques using pinhole-free cathodic coating8.
Based on the observation that the anti-corrosion performance of Cu-FA depends on the formate treatment time (Extended Data Fig. 3a, b), we investigated the underlying mechanism. Although switching from the formate–H2O system to the formate–oleylamine system in mixed DMF–H2O decreased the temperature from 200 °C to 160 °C, a long reaction time was still required to achieve effective passivation. Surprisingly, regardless of which treatment system was used and which original crystalline orientation of the polycrystalline Cu foil was selected, the relative intensity of the Cu(110) diffraction peak increased with treatment time, indicating that deep lattice reconstruction of the Cu surface took place during the formate treatment. Cross-sectional samples of the Cu foils before and after the formate treatment were prepared by the focused-ion-beam technique for the direct imaging of lattice changes using transmission electron microscopy (TEM) (Extended Data Fig. 3c, d). Whereas the surface of the untreated Cu foil was mainly Cu(111), the surface of the formate-treated Cu foil was reconstructed to Cu(110), which was not limited to surface atomic layers, but extended to a thickness of over 200 nm.
To identify the atomic structure of the surface passivation layer on Cu, we carried out combined scanning tunnelling microscopy (STM) and atomic-force microscopy (AFM) experiments using a qPlus sensor17,18. Contaminants on the Cu-FA foils were removed by annealing in an ultrahigh vacuum (UHV) chamber (Extended Data Fig. 4a). Although annealing at 150 °C already gave STM images showing step planes and sharp step edges, it was impossible to obtain high-resolution STM images. Increasing the annealing temperature to 300 °C produced un-contaminated surfaces for high-resolution imaging. Although microcrystalline Cu foils were used for the formate treatment, large-scale single-crystalline domains (Fig. 2a) were observed on Cu-FA. In comparison, untreated Cu foils showed poorly defined rough surfaces after the same annealing. The steps on Cu-FA were uniform with a height of about 2.55 Å, corresponding to the diatomic step height of Cu(110), indicating that the microcrystalline Cu surface was mostly reconstructed to Cu(110) after the formate treatment. The same step heights were observed for the samples annealed at 150 °C and 300 °C, confirming that the reconstruction was not due to the annealing at 300 °C, but induced by the formate treatment, as suggested by XRD and TEM.

a, STM topography of the formate-treated Cu foil. Inset, line profile across an atomic step. Before imaging, the sample was annealed under UHV at 300 °C. b, Zoom-in STM image showing a c(6 × 2) superlattice. The unit cell is highlighted by a white rectangle. Set points of STM images: 1 V, 20 pA (a); −1 V, 50 pA (b). c, High-resolution constant-height AFM image (Δf, frequency shift). The \([1\,\bar{1}\,0]\) row of Cu(110) is highlighted by a red arrow. Inset, zoom-in AFM image of the paired triangular lobes (top) and corresponding structural model (bottom). The AFM image was recorded with a tip height of −0.3 nm, referenced to the STM set point (1 V, 5 pA). The oscillation amplitude is 0.1 nm. d, Simulated AFM image based on the DFT structure shown in e, using a neutral-tip model (effective lateral stiffness k = 0.25 N m−1, oscillation amplitude A = 0.1 nm). e, Dehydrated structure model of Cu(110) passivated by dinuclear Cu(ii)–formate motifs. The structure was built by removing H2O from the structure model of a Cu(110)-c(6 × 2) surface fully passivated by [Cu(μ-HCOO)(OH)2]2 and O2− (Extended Data Fig. 4c). Blue spheres, Cu on the surface; green spheres, Cu on the subsurface; light-grey spheres, Cu in the bulk; white spheres, H; red spheres, O; grey spheres, C.
From the high-resolution STM images of Cu-FA, a perfect Cu(110)-c(6 × 2) superlattice (Fig. 2b) was identified. It should be noted that the c(6 × 2) superstructure (Extended Data Fig. 4b) was also observed on single-crystal Cu(110) treated with sodium formate solution, followed by annealing at 150 °C; this superstructure was comparable to that created by the formate treatment in providing effective passivation, implying that it did not result from annealing at 300 °C. In the constant-height AFM image, each round protrusion in the STM image was further resolved to be paired non-spherical lobes (Fig. 2c). In addition, the \([1\,\bar{1}\,0]\) rows of the underlying Cu(110) are visible, as indicated by the red arrow in Fig. 2c. In the zoom-in AFM image, the paired lobes have a distinct triangular shape with a separation of 3.73 ± 0.03 Å (standard deviation of four measurements) (inset of Fig. 2c), which is larger than the distance between neighbouring Cu atoms (2.5 Å) on the \([1\,\bar{1}\,0]\) row, precluding that the bright protrusions originate from bidentate formates on the \([1\,\bar{1}\,0]\) row19.
Cu easily forms paddle-wheel dinuclear Cu(ii) carboxylate coordination compounds20. When the ligands are formates, the distance of C atoms on each pair of perpendicularly bridging formates is typically in the range 3.2–4.0 Å (Extended Data Fig. 4c). Moreover, the presence of bridging formates on Cu-FA annealed under similar conditions as those used for STM imaging was confirmed by Fourier transform infrared (FTIR), Raman spectroscopy and temperature-programmed desorption–mass spectrometry (TPD-MS) studies (Extended Data Fig. 4d). With the presence of formates, the Cu-FA foils annealed in UHV still exhibited the same anti-corrosion performance as before annealing (Extended Data Fig. 4e). On the basis of these results, a (6 × 2) supercell of Cu(110) with a five-layer-thickness slab was built for periodic density functional theory (DFT) calculations using the Perdew–Burke–Ernzerhof (PBE) functional as implemented in the Vienna ab initio simulation package21. The slab was coated by a layer consisting of dinuclear [Cu(μ-HCOO)(OH)2]2 units and O2− arranged in a c(6 × 2) configuration (Extended Data Fig. 4c). Although different Cu(110) structures modified with formate have been extensively investigated22,23,24,25, a structure modified with dinuclear Cu–formate motifs has not yet been reported in the literature. This structure can be viewed as a gradual material with the formal valence of Cu decreased from +2 on the surface dinuclear Cu motifs to +1 in the subsurface and to 0 in the inner Cu atoms, as revealed by Bader charge analysis and confirmed by linear sweep voltammetry and by Auger and X-ray photoelectron (XPS) spectroscopies (Extended Data Fig. 4f-g).
XPS measurements also suggested that a dehydration process occurred on Cu-FA upon annealing under conditions (UHV, 300 °C) similar to those of the pretreatment for the high-resolution AFM/STM imaging. Whereas the presence of OH− was observed for the unannealed sample, the annealed Cu-FA displayed XPS signals of O from the formate and O2− on Cu, but no obvious OH− signal. According to the observed dehydration process, the dehydrated structure model (Fig. 2e) was optimized by DFT calculations. The simulated AFM image of the optimized structure (Fig. 2d) shows not only the carbon–carbon distance of 3.66 Å between the two coordinated formates, but also the fine triangular structure of the formate moieties, consistent with the experimental observations (Fig. 2c). The dehydrated c(6 × 2) structure should therefore be the structure observed by STM after annealing at 300 °C. However, once the dehydrated structure was exposed to the atmosphere, the occurrence of the hydration process was revealed by STM (Extended Data Fig. 5a), with the appearance of defect-like dark depressions, which are attributed to interstitial O2− species on the hydrated c(6 × 2) structure (Extended Data Fig. 4c). Therefore, the hydrated c(6 × 2) structure must be the real passivating structure, although the dehydrated structure is easily obtained upon annealing (Extended Data Fig. 5b).
The passivation layer reveals the two important features. (1) Whereas the valence of the outmost surface Cu is +2, that of the subsurface Cu atoms is +1. All exposed Cu sites are already in the oxidized state. (2) The subsurface Cu atoms are fully bound by O2− and OH− groups. In alkaline conditions, the binding of OH− on oxidized Cu species is so strong that, for example, Cu(OH)2 has a very small solubility product constant of 2 × 10−22. These features make it difficult for O2 or other adsorbates to interact with Cu underneath the passivation layer. The perfect symmetry match between Cu(110) and the four OH− binding sites on [Cu(μ-HCOO)(OH)2]2 is crucial to the passivation. It is therefore critical to have the Cu surface reconstructed into Cu(110) before a robust passivation layer can be created, which explains why a long treatment time was required to achieve effective anti-corrosion. As expected, when a single-crystal Cu(110) surface was treated with formate solution at 100 °C (Fig. 3a–c, Extended Data Fig. 5c, d), an anti-corrosion effect was readily achieved, even when the treatment time was reduced to 1 h. In comparison, no anti-corrosion protection was identified on Cu with (111) and (100) surfaces, even by extending the treatment to 10 h under the same conditions. However, when the Cu(111) surface was scratched to introduce defect sites, the reconstruction of the single-crystal surface was readily accelerated to give anti-corrosion protection (Extended Data Fig. 5e), suggesting that the reconstruction must be initiated at the domain boundaries. These results confirmed the importance of rearranging the Cu surface into Cu(110) before the formate passivation becomes effective.

a–c, SEM images of formate-treated single-crystalline surfaces after exposure to 0.1 M sodium hydroxide for 12 h: Cu(110)-FA (a), Cu(100)-FA (b) and Cu(111)-FA (c). d, Comparison of adsorption free energies of O2 and Cl− at 298 K on a clean Cu(110) surface and on Cu(110)-FA with surface co-passivation by [Cu(μ-HCOO)(OH)2]2 and O2−. e, Comparison of the mass of dissolved Cu(ii) from Cu-FA/DT, Cu-FA and Cu after the salt spray test in 5% NaCl at 47 °C for 96 h. The data are averages of three independent measurements. Error bars represent the standard errors. f, Optical photographs of Cu-FA/DT (1), Cu-FA (2) and Cu (3) foils after a 10-min exposure to Na2S (50 mM).
As demonstrated by DFT calculations (Fig. 3d, Extended Data Fig. 5f), whereas O2 binds strongly with clean Cu(110), the Cu(110)-c(6 × 2) surface passivated by [Cu(μ-HCOO)2(OH)2]2 and O2− leaves no accessible sites for O2 to be adsorbed and activated so that the oxidation of Cu can be switched off. DFT calculations also suggested that the anti-corrosion properties of Cu-FA surfaces should be enhanced if very strong alkanethiol ligands are used to coordinate with steps or defect sites that are not protected by [Cu(μ-HCOO)(OH)2]2 units (Extended Data Fig. 6a). 1-dodecathiol (DT) was therefore chosen for the demonstration. The Cu-FA foil that was further treated with DT (denoted as Cu-FA/DT) exhibited a substantially enhanced anti-corrosion performance (Fig. 3e, f, Extended Data Fig. 6b–g) with a corrosion rate of 0.755 μm yr−1 in 0.1 M NaOH, two orders of magnitude lower than that of bare Cu (78.2 μm yr−1). The surface of Cu-FA/DT remained almost intact after exposure to salt spray for 24 h. Moreover, compared with Cu-FA, the Cu-FA/DT foil increased its Na2S-resistant concentration by two orders of magnitude. No obvious formation of dark copper sulfide was observed on Cu-FA/DT after 5 h of immersion in 10 mM Na2S. Cu-FA/DT also exhibited enhanced oxidation resistance in 720-h seawater inundation and 30% H2O2.
Understanding of this mechanism provided an alternative strategy to achieve effective passivation by introducing Cu(HCOO)2 as the metal precursor for the reductive growth of a Cu(110) surface on Cu foils. This strategy allows us to create well passivated a Cu(110) surface at 120 °C, making the technique applicable to Cu materials of different forms and sizes (Extended Data Fig. 7). For example, the electric conductivity of treated Cu wires was well maintained even after 60 h of ageing in 0.1 M NaOH at 60 °C. No obvious surface change was observed for treated Cu wires subjected to 24 h of air oxidation at 160 °C. The Cu-FA/DT meshes were highly corrosion-resistant to salt spray and Na2S. Moreover, no obvious colour change was observed on Cu-FA/DT tubes and foils after a 96-h wet mechanical test in which salty water (3.5% NaCl, 1% Na2CO3, 1% Na2SO4 and 0.1% NaOH) was running through the samples intermittently, suggesting good wet mechanical properties for the passivation layer.
The developed anti-corrosion protocol also works well for Cu nanomaterials (Extended Data Fig. 8). Cu nanowires (NWs) with an average diameter of ~35 nm and length of ~220 μm were synthesized for the assessment26. The unmodified Cu NWs in solution were unstable in air and were easily oxidized to form a surface oxide layer, thus dramatically reducing the electronic conductivity27. After the formate treatment, the Cu NWs (denoted as Cu NWs-FA) showed high stability in air for 90 days owing to their fully passivated Cu(110) surface. No formation of Cu oxide species was detected from Cu NWs-FA after 48 h of thermal treatment at 80 °C in air (Fig. 4a–d). However, because of their high surface area, the Cu NWs-FA did not survive under harsh alkaline corrosion conditions of 1 M NaOH in air. When further treated with DT, the resulting Cu NWs-FA/DT exhibited an enhanced anti-corrosion performance under harsh conditions (that is, 1 M NaOH in air, salt spray at 47 °C), which makes them a good candidate for fabricating transparent conductive electrodes. Using Cu NWs-FA/DT, we obtained a transparent conductive electrode with sheet resistance of 25.8 Ω per square (light transmission of 87% at 550 nm) and with good air and mechanical stabilities (Extended Data Table 1). Highly corrosion-resistant Cu nanoparticles were also prepared using the developed protocol. Moreover, the technique was successfully applied to kilogram-scale production of Cu pastes under mild conditions to replace Ag pastes for radiofrequency identification applications (Extended Data Fig. 9a–d).

a–c, SEM images of formate-treated Cu NWs (Cu NWs-FA) (a) and untreated Cu NWs (b) after 48-h air oxidation at 80 °C, and their corresponding XRD patterns (c). Numbers in parentheses show the powder diffraction file card numbers of the samples (JCPDS, joint committee on powder diffraction standards). d, Resistance change profile of Cu NWs and Cu NWs-FA with heating time at 80 °C. e, XRD pattern of Cu foil before and after (Cu-FA(EC)) electrochemical treatment in the presence of sodium formate. f, Optical photographs of a Cu foil obtained by roll-to-roll electrochemical treatment followed by thiol treatment (Cu-FA(EC)/DT) and untreated Cu foil after ageing in 50 mM Na2S for 10 min.
The importance of the Cu anti-corrosion technique reported here is supported by the development of a facile, quick and efficient electrochemical process to fabricate anti-corrosive Cu surface at ambient conditions (Extended Data Fig. 9e–i). Lattice reconstruction for Cu surfaces under electrochemical redox conditions has been well documented28,29,30. In our electrochemical process, an anodic sweep from −0.80 V to 0.20 V (versus a saturated calomel electrode, SCE) in an electrolyte containing 1.0% HCOONa (pH 8–10) at a scan rate of 10 mV s−1 was applied to generate oxidized Cu species, followed by a 5-min electrochemical reduction at −0.7 V (versus SCE), during which the cathodic reduction of Cu(i) into Cu(0) occurred. As illustrated in Fig. 4e, the diffraction peak of Cu(220) was obviously enhanced after the electrochemical treatment, suggesting that the electrochemical treatment also induced reconstruction similar to that achieved by the solvothermal method. The as-prepared Cu-FA(EC) foil also exhibited identical anti-corrosion properties to the Cu-FA foils made by the solvothermal method. Similarly, further DT treatment can enhance the performance to make Cu highly corrosion-tolerant to Na2S. The electrochemical method developed in this work makes it possible to continuously produce anti-corrosive Cu materials using roll-to-roll processes for practical applications (Fig. 4f). However, we should point out that in the application of the formate technique, acidic conditions with pH < 3 must be avoided, because the formate passivation layer cannot survive under such conditions.
Methods
Materials
The Cu foil (99.8%, metal basis, thickness 25 μm, No. 046986) and polyvinylpyrrolidone (PVP; molecular weight 55,000) were purchased from Alfa Aesar Chemical Reagent Co. Ltd (Tianjin, China). Cu(111), Cu(110) and Cu(100) single crystals were obtained from Hefei Kejing Materials Technology Co., Ltd (Hefei, China). 1-DT (analytical reagent, AR), CuCl2·2H2O (AR), glucose (AR), oleylamine (80−90%) and oleic acid (90%) were purchased from Aladdin Reagent Co., Ltd (Shanghai, China). Sodium formate (HCOONa), sodium sulfide (Na2S), copper sulfate (CuSO4·5H2O), sodium hydroxide (NaOH), diethyldithiocarbamate (DDTC), sodium ethylenediamine tetraacetate (EDTA), ammonium citrate, ethylene glycol (EG), DMF, carbon tetrachloride (CCl4), acetone, ethanol and other reagents were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Brass foil (H68), bronze foil, Cu wire (99.9%, diameter 2 mm) and Cu mesh (99.9%, thickness 50 μm) were obtained from Shenzhen Xinming Metal Material Co., Ltd (Shenzhen, China). All reagents were used as received without further purification. The water used in the studies was ultrapure water with a resistivity of ≥18 MΩ cm provided by a Millipore water purification system (Bedford, USA).
Hydrothermal and solvothermal formate treatments of copper (Cu-FA)
The following three methods were developed to treat Cu surfaces with formate for effective anti-oxidation passivation.
Method I
Hydrothermal treatment of Cu in the presence of formate. In a typical treatment, a 2 cm × 2 cm × 0.025 mm Cu foil or mesh or a 10-cm-long Cu wire is placed with 500 mg HCOONa in 10 ml of nitrogen-saturated H2O. The mixture is sealed in a 50-ml stainless-steel autoclave. The mixture is then heated from room temperature to 200 °C in 40 min and kept at 200 °C for 24 h to prepare Cu-FA. The Cu-FA products are collected and rinsed with deionized water to remove free HCOONa.
Method II
Solvothermal formate treatment of Cu in the presence of oleylamine (Cu-FA). In a typical treatment, a 2 cm × 2 cm × 0.025 mm Cu foil or mesh or a 10-cm-long Cu wire and 200 mg HCOONa in 1 ml H2O are dissolved in 12 ml DMF by sonication. Then, 1 ml oleylamine is added into the mixture and sonicated for another 3 min. The mixture is sealed in a 50-ml stainless-steel autoclave. The mixture is then heated from room temperature to 160 °C in 30 min and kept at 160 °C for 16 h to prepare Cu-FA. The Cu-FA products are collected and rinsed three times with deionized water and ethanol alternately to remove free HCOONa and oleylamine.
Method III
Solvothermal formate treatment of Cu in the presence of oleylamine and Cu(HCOO)2. In a typical treatment, a 2 cm × 2 cm × 0.025 mm Cu foil or mesh or a 10-cm-long Cu wire, 0.1 mg Cu(HCOO)2·4H2O and 200 mg HCOONa in 1 ml H2O are mixed in 12 ml DMF by sonication. Then, 1 ml oleylamine is added into the mixture and sonicated for another 3 min. The mixture is sealed in a 50-ml stainless-steel autoclave. The mixture is then heated from room temperature to 120 °C in 20 min and kept at 120 °C for 12 h to prepare Cu-FA. The Cu-FA products are collected and rinsed three times using deionized water and ethanol alternately to remove free HCOONa and oleylamine.
Formate treatments of Cu single crystals
In a typical treatment, two species of 1 cm × 1 cm × 0.5 cm Cu single crystals with exposure surfaces of Cu(110), Cu(100) or Cu(111) were placed in 10 ml H2O containing 200 mg HCOONa (N2-purged). The mixture was sealed in a 50-ml stainless-steel autoclave. The sample was then heated from room temperature to 100 °C in 20 min and kept at 100 °C for different times to prepare Cu(hkl)-FA. The single crystals were collected and rinsed with deionized water to remove free HCOONa.
Preparation of copper foils, meshes and wires co-stabilized by formate and 1-DT (Cu-FA/DT)
The as-prepared Cu-FA samples were dried in vacuum before use. In order to prepare Cu-FA/DT, the Cu-FA samples were immersed in 20 ml 1 mM DT/EtOH solution and kept still for 5 min, and were then washed with pure ethanol three times to produce Cu-FA/DT.
Preparation of graphene-coated Cu foils (Cu-G)
The Cu-G samples were grown by the chemical vapour deposition (CVD) method31. Cu foils (25 μm thick, 99.8%, Alfa Aesar) were placed on a flat oxide substrate and then loaded into a CVD system (Hefei Kejing Company, OTF1250). The system was heated to 1,000 °C in 1 h with Ar (500 standard cubic centimetres per minute, sccm), followed by annealing in additional H2 (10 sccm) for 40 min. Then, CH4 gas (0.1 sccm) was introduced as the carbon source for graphene growth. Finally, the system was cooled naturally with Ar (500 sccm) and H2 (10 sccm).
Preparation of Cu NWs
In a typical synthesis of Cu NWs32, CuCl2·2H2O (680 mg, 4 mmol) and glucose (792 mg, 4 mmol) were dissolved in 80 ml of deionized water by stirring in a beaker. In another beaker, 8 ml oleylamine and 80 μl oleic acid were dissolved in ethanol by stirring. Afterwards, these two solutions were put into a 500-ml beaker and diluted to 400 ml with water, followed by magnetic stirring for 16 h at 50 °C. After the colour of the mixture changed from blue to cyanish grey, the mixture was transferred to a stainless-steel autoclave and then heated from room temperature to 120 °C in 20 min. After heating at 120 °C for 6 h, the mixture was cooled to room temperature. The upper yellow suspension was discarded and the bottom reddish-brown Cu NWs were obtained. The Cu NWs were then centrifuged and washed with a 1:1 ethanol:water mixture (100 ml, 8,000 rpm, 2 min) and pure ethanol. Finally, the purified Cu NWs were dispersed into 100 ml ethanol to obtain a Cu–nanowire dispersion with a content of ~1 mg ml−1.
Preparation of Cu NWs-FA and Cu NWs-FA/DT
In a typical synthesis, 10 mg Cu NWs, 0.1 mg Cu(HCOO)2·4H2O and 200 mg HCOONa in 1 ml H2O were dissolved in 12 ml EG by sonication to form a homogeneous solution. Then, 1 ml oleylamine was added into the mixture and sonicated for another 3 min. The mixture was sealed in a 50-ml stainless-steel autoclave, and was heated from room temperature to 120 °C in 20 min and kept at 120 °C for 12 h to prepare Cu NWs-FA. Following the same protocol, when the EG was replaced by 12 ml DMF, Cu NWs-FA were also readily prepared by heating the mixture from room temperature to 160 °C in 20 min and then maintaining it at 160 °C for 16 h. The Cu NWs-FA products were collected and rinsed three times with ethanol and deionized water alternately to remove free HCOONa and oleylamine. The as-prepared Cu NWs-FA was dried in vacuum before use. To prepare Cu NWs-FA/DT, 10 mg of the as-prepared Cu NWs-FA was dispersed in 20 ml EtOH containing 1 mM DT by magnetic stirring for 10 min and was then centrifuged with a 1:1 ethanol:water mixture (100 ml, 8,000 rpm, 2 min) and pure ethanol.
Preparation of Cu nanoparticles (Cu NPs)
Cu NPs were synthesized using a previously reported method33. 11.1 g PVP (K30) and 4 g sodium hypophosphite were mixed into 40 ml EG inside a round-bottom flask while vigorously stirring at room temperature under ambient atmosphere. The mixture was heated to 90 °C at a rate of 5 °C min−1. Then, 10 ml of a 1 M solution of copper sulfate (CuSO4·5H2O) in EG at 90 °C was rapidly added into the PVP–sodium hypophosphite solution while stirring vigorously. As reduction occurred, the colour of the suspension turned from green to reddish brown within 2 min, indicating the formation of Cu NPs. The reaction was quenched, and the suspension was rapidly cooled by adding chilled deionized water. The Cu NPs were separated and washed with deionized water by centrifugation, and acetone was used as a non-solvent to remove excess PVP and side products. The resulting precipitates were dried under vacuum at 60 °C for 2–3 h before formate decoration.
Preparation of FA-treated Cu NPs (Cu NPs-FA)
In a typical synthesis, 10 mg Cu NPs, 0.1 mg Cu(HCOO)2·4H2O and 200 mg HCOONa in 1 ml H2O were dissolved in 10 ml EG by sonication to form a homogeneous solution. Then, 1 ml oleylamine was added into the mixture and sonicated for another 3 min. The mixture was sealed in a 50-ml stainless-steel autoclave, and was then heated from room temperature to 120 °C in 20 min and kept at 120 °C for 12 h to prepare Cu NPs-FA. The Cu NPs-FA products were collected and rinsed three times with ethanol and deionized water alternately to remove free HCOONa and oleylamine. The as-prepared Cu NPs-FA was dried in vacuum before use.
Preparation of formate-coated copper foil by the electrochemical method (Cu foil-FA-EC)
The Cu foil-FA-EC was investigated on a CHI 760E electrochemical workstation in a three-electrode configuration cell using polished Cu foil as the working electrode, a platinum plate (1 × 1 cm2) as the counter electrode and an SCE as the reference electrode in 1% HCOONa/NaOH aqueous electrolyte (pH 8–10), whereas the active area was 1 cm2. Cyclic voltammetry curves were collected in the potential window of −800 mV to 200 mV at a scan rate of 10 mV s−1. Electrochemical reconstruction was achieved by the chronoamperometry method, in which the applied potential is the redox potential of Cu(i) to Cu(0).
Electrochemical corrosion measurements
To quantitatively characterize the corrosion rates of Cu foils treated with different methods, electrochemical measurements were conducted with a standard three-electrode configuration in a 0.1 M NaOH or 1 M NaOH (Sigma Aldrich) solution at room temperature. The following Cu samples were compared: bare Cu foil, Cu-FA, Cu-FA/DT, Cu-BTA and Cu-DT. In all measurements, Cu foils with an area of 2 cm2 were used as the working electrode, a Pt wire was used as the counter electrode and Ag/AgCl was used as the reference electrode. All electrochemical measurements were carried out using a potentiostat with a voltage sweep rate of 20 mV s−1. The corrosion rates (CR) of the samples were determined by the kinetics of both anodic and cathodic reactions34 and calculated from the corrosion current density, Jcorr, as:
where K = 3,272 mm A−1 cm−1 yr−1 is the corrosion rate constant, and EW and ρ are the equivalent weight and mass density of the corroding species, respectively.
Electrochemical imaging with scanning reference electrode
Potential variation images of the sample surface were recorded by an integrated SRET/STM system (Xiamen Le Gang Materials Technology Co. Ltd, China)35,36, which can simultaneously map the potential or current distribution and the topography on metal surfaces. The SRET/STM operates at SRET and STM scanning modes using a reference Pt–Ir probe and a local reference Pt–Ir probe, respectively. The sample is connected to the ground, the reference probe measures the average electrochemical potential (Eaverage) in bulk solution, whereas the local reference probe detects the local electrochemical potential (Elocal) near the sample surface, which is strongly influenced by local electrochemical reactions taking place on the sample surface. In SRET mode, the recorded potential signal is the potential difference ∆E between the reference and local reference probes, ∆E = Eaverage − Elocal. The two signals (Elocal and Eaverage) are fed into a differential electrometer to generate the resulting potential signal (∆E) to construct three-dimensional current images. As a result, local anodes have a high ∆E in the potential or current images.
Stability tests under different environments
Stability in alkaline solutions: for the alkaline-resistance test, the Cu materials were immersed in 0.1 M NaOH solutions at room temperature for different intervals. For the corrosion rate evaluation, the Cu materials were immersed in 0.1 M NaOH solutions under different voltages for electrochemical tests. Stability in salt spray conditions: the Cu materials were placed in a HD-E808-60 Salt Spray Test Chamber (Haida Instruments Co. Ltd, China) with 5% NaCl and relative humidity >100% at 47 °C. Stability in Na2S solution: the Cu materials were immersed in Na2S solutions of different concentrations (1 mM to 1 M) at room temperature for 0–120 min for the Na2S-resistance test. Stability in H2O2 solutions: the Cu materials were immersed in H2O2 of different concentrations for the anti-oxidation evaluation. Stability in air at 80 °C and 80% relative humidity: freshly made samples were placed in air at 80 °C and relative humidity of (80 ± 5)%. Stability under wet mechanical conditions: salty water (3.5% NaCl, 1% Na2CO3, 1% Na2SO4 and 0.1% NaOH) was continuously supplied through the tubing samples (inner diameter 1.6 cm) at a flow rate of 1,400 l h−1 or intermittently through foil samples at an overall flow rate of 400–800 l h−1.
Characterizations
Raman spectra were obtained on XploRA (Jobin Yvon-Horiba, France) confocal Raman microscopes with a dark-field function. The excitation wavelength was 532 nm from a He–Ne laser and the power on the sample was about 1 mW. We used a 50× magnification long-working-distance (8 mm) objective to focus the laser onto the sample and collect the backscattered light.
Optical microscope images of the surfaces of different Cu samples were recorded on an Olympus BH2-UMA optical microscope in reflectance mode with a Moticam 2000 2.0 M pixel camera, on a Nikon Eclipse Ti–U optical microscope in reflectance mode with an Ample Scientific 3.0 M pixel camera or on the confocal Raman microscopes.
SEM measurements were carried out on Zeiss SIGMA microscope at an accelerating voltage of 15 kV. The powder XRD experiments were conducted on a Rigaku Ultima IV XRD system using Cu Kα radiation. The operation voltage and current were 40 kV and 30 mA, respectively. The scanning speed was 10° min−1. In order to obtain the surface reconstruction information, a grazing-incidence XRD mode was used.
TEM studies were performed on a TECNAI F30 TEM operating at 300 kV. The samples were prepared by dropping an ethanol dispersion of the samples onto 300-mesh carbon-coated copper grids and immediately evaporating the solvent. High resolution-TEM and high resolution-STEM were performed on an atomic-resolution analytical microscope (JEM-ARM 200F) operating at 200 kV. The Cu NWs and Cu NWs-FA/DT were prepared by dropping an ethanol or cyclohexane dispersion of the samples onto 300-mesh carbon-coated copper grids and immediately evaporating the solvent. To further confirm the surface reconstruction, the focused-ion-beam technique was used to prepare the cross-sectional TEM samples of the Cu foil surfaces before and after the formate treatment for characterizing their surface layer structures using JEM-ARM 200F.
The infrared spectra of the solid samples were recorded from 4,000 cm−1 to 650 cm−1 on a Nicolet iS10 FTIR spectrometer (Thermo Scientific Corporation) in the attenuated total reflection–FTIR mode.
XPS and X-ray-induced Auger electron spectroscopy analyses were performed using a Thermo Fisher Scientific ESCALAB 250Xi spectrometer with focused monochromatic Al Kα radiation (1,486.6 eV; 150 W; 500 μm diameter of irradiated area). A hemispherical electrostatic analyser and a standard lens were used to maximize the signal. The basic pressure inside the analysis chamber was about 5 × 10−10 mbar. The takeoff angle between the direction of the analyser and the Cu foil was ±45°. The data were averaged over more than 10 scans during the measurement and were used without further smoothing. Peak fitting was processed using CasaXPS software. A Shirley-type background subtraction was performed before curve fitting, and the experimental curve for the Cu 2p3/2 peak fitting was set to a mixed Gaussian and Lorentzian function (defined in CasaXPS as GL(90))37.
TPD-MS was performed on a home-built TPD time-of-flight (TOF) analyser. 10.0 mg of Cu NWs or 200 mg of Cu wire, foil or mesh were pyrolyzed in a small tube heated by a heating coil. A K-type thermocouple was put inside the sample tube and insulated from the samples to measure the temperature. The heating coil was powered by a precise electric source and adjusted in intervals of 10 mV. The temperature of the sample tube was ramped smoothly from room temperature to 800 °C at a rate of 5 °C min−1, controlled by a computer. The desorbed species were ionized by an ultraviolet lamp positioned very close to the sample tube with a photon energy of 10.6 eV, and then transferred to the TOF analyser by an ion optical system. The TOF analyser had a resolution of over 5,000 and a sensitivity of the order of parts per billion. All these steps were performed in high vacuum (~3 × 10−5 Pa). The mass spectrum and sample temperature were recorded every second. Each spectrum is an accumulation of 10,000 spectra gathered at intervals of 100 μs.
The resistivity of the Cu samples was measured on a CHI 760E electrochemical workstation. The resistances of the copper foils were measured using a four-probe method (ST2263, Suzhou Jingge Electronic Co. Ltd). For the Cu wires, the electric conductivity (σ) is calculated according to the equation σ = L/(R × Sa), where L is the length of the Cu wire, Sa represents the cross-sectional area and R is the resistance. For the copper foil or films, the square resistance was acquired and σ = 1/(sheet resistance × thickness of the foils or films).
The thermal diffusivity (α; in m2 s−1) of the Cu foil was measured on an LFA Nanoflash 467 Light flash system. The in-plane thermal diffusivities were measured for the Cu foil and Cu foil-FA samples before and after a corrosion test with 0.1 M NaOH. The thermal conductivity (κ) is calculated according to the equation κ = α × ρ × Cp, with apparent density ρ = 8.96 g cm−3 and specific heat capacity Cp = 0.39 × 103 J kg−1 K−1.
The contact angles of different Cu samples were measured using a Ramé–Hart M500 digital goniometer equipped with a dispensing needle (VICI Precision Sampling, CA, USA). A 2-μl water droplet was generated using the automatic dispenser of the goniometer. The sessile droplet was formed by fixing the needle and approaching the substrate parallel to the needle direction with a gentle feed rate of a few micrometres per minute. All the tests were carried out in air at room temperature. The axisymmetric drop-shape analysis profile method was used to estimate the contact angle of the water droplet on the solid surface.
STM and AFM measurements
STM/AFM experiments were performed with a combined AFM/STM system (Createc, Germany) at 5 K with base pressure <5 × 10−9 Pa. Electrochemically etched Pt–Ir tips were sharpened by a focused ion beam and cleaned by alternate annealing and sputtering before the experiments, and further controlled by field-emission and voltage-pulse procedures during the scanning. Before the STM investigations, the samples were annealed in vacuum by heating up to 300 °C for an extended time (typically longer than 10 h) in a pre-chamber with base pressure <1.5 × 10−8 Pa. The Cu(110) single-crystal sample with the c(6 × 2) structure was obtained in UHV with the following procedure. 1) The Cu(110) single crystal was cleaned by five sputtering–annealing cycles. 2) The clean Cu(110) sample was quickly transferred from the UHV chamber to a load-lock chamber protected by N2 gas, where a droplet of ultrapure sodium formate solution was dripped onto the Cu(110) surface. 3) The Cu(110) sample covered by formate solution was transferred back to the UHV chamber, followed by low-temperature annealing (100 °C–300 °C) to obtain the c(6 × 2) structure. Bias voltage refers to the sample voltage with respect to the tip. All the STM topographic images were obtained in constant-current mode and the AFM images in constant-height mode.
Simulations of AFM images
The Δf images were simulated with a molecular mechanics model including the electrostatic force, according to the methods described in refs. 38,39. We used the following parameters in the flexible probe–particle tip model: effective lateral stiffness k = 0.25 N m−1 and effective atomic radius Rc = 1.748 Å. The parameters of the Lennard–Jones pairwise potential for all elements were taken from the force field developed by Zhao et al.40.
Quantification of Cu(ii) ions
The amount of Cu(ii) ions was quantified by the standard sodium diethydlthiocabamate spectrophotometric method (China Standard HJ 485-2009). In ammonia solution (pH 8-10), Cu(ii) reacts with DDTC to produce a yellow–brown complex. The Cu(ii)-DDTC complex can be extracted using CCl4. The absorbance is measured at a wavelength of 440 nm and the colour is stabilized for 1 h. Iron, manganese, nickel and cobalt react with DDTC to form coloured complexes, which can be masked by EDTA-ammonium citrate solution to eliminate interference.
Computational details
Spin-polarization calculations were carried out at the PBE level using the Vienna ab initio simulation package (VASP 5.3.5). The valence electrons were described by plane-wave basis sets with a cutoff energy of 400 eV, and the core electrons were replaced by projector-augmented wave pseudopotentials41,42,43,44,45.
It should be noted that different Cu(110) structures modified with formate species have been extensively investigated using both experiments and calculations23,24,25,46,47,48. Although the formate species that preferentially adsorb on Cu(110) have been well documented, the formate coordination inducing the reconstruction of Cu surfaces into Cu(110) has not been studied. In our DFT simulations, we adopted the c(6 × 2) structure with dinuclear [Cu(μ-HCOO)(OH)2]2 units on the Cu(110) surface. Accordingly, a (6 × 2) supercell with five-layer-thickness slabs was built. The vacuum regions between the slabs were set as 15 Å and k-point sampling was performed following the Monkhorst–Pack procedure with a 3 × 2 × 1 mesh49. During structural optimization, the bottom two layers of the slab were fixed at a bulk truncated position while the top three layers and the adsorbates were allowed to be fully relaxed. All internal structural parameters were allowed to relax until the Hellman–Feynman forces on each ion were lower than 0.02 eV Å−1.
The adsorption energies (ΔEads) were calculated using equation (2), where Ead/sub, Ead and Esub are the total energies of the optimized adsorbate/substrate system, the adsorbate and the clean substrate, respectively.
Because the triplet O2 was poorly described by generalized gradient approximation functionals, we used gas-phase H2O and H2 as references to estimate the total energy of O2 using equation (3).
Here, ΔEr denotes the reaction heat at 0 K without a zero-point energy correction, which can be deduced from the experimental atomic energies of O2, H2 and H2O using equation (4).
where AE(H2O), AE(H2) and AE(O2) represent the atomic energies of H2O, H2 and O2, respectively. Our calculations showed that the corrected total energy of O2 was −9.41 eV.
Here, we neglected the zero-point energy and thermal correction, and used equation (5) to estimate the Gibbs free energy of O2 adsorption at 298 K (ΔGads). We assumed that adsorption would lose translational, rotational and vibrational entropies50 and adopted the experimental standard entropy (ΔS) of gaseous O2 (205.15 J K−1 mol−1).
Similarly, we estimate the adsorption free energy of Cl− at 298 K as
where ΔEads(Cl−) is the adsorption energy of the gaseous Cl atom, taken from DFT calculations; EA, Φ and ΔGsol(Cl−) represent the electron affinity of Cl atom (−3.61 eV), the work function of Cu(110) (−4.48 eV) and the solvation free energy of Cl− (−3.23 eV), respectively, taken from experimental values51. Considering that the gaseous Cl− has only three-dimensional translational freedom, ΔS (Cl−, gas) could be calculated using the Sackur–Tetrode formula50,52,
where m is the molecular mass of Cl−, V/N is the volume per molecule in the standard state, kB is the Boltzmann constant, Rg is the gas constant and h is the Planck constant. From equation (7), the translational entropy of Cl− in the gas phase was calculated to be 153 J mol−1 K−1.
To identify the oxidation states of surface Cu, we carried out Bader charge analysis. In terms of molecular orbital theory, the wavefunction belongs to the whole system, and not to individual atoms. Bader’s theory of ‘atoms in molecules’53 provides a reliable scheme to partition the electronic charge density between the atoms according to zero-flux surfaces. It should be noted that the Bader charge is not the same as the oxidation state, but there are a few situations in which the two are correlated. To determine the oxidation state of an element (such as Cu), the Bader charges of a series of oxides of the element in different oxidation states (such as metallic Cu, Cu2O and CuO) are calculated first and used as references to calibrate the oxidation state of the element in an unknown system54,55.
Transparent conductive films based on Cu NWs
To make a transparent conductive thin film, a dilute suspension of Cu NWs in toluene was produced using sonication. A thin film was then fabricated by filtering down the nanowire suspensions onto a nitrocellulose porous membrane (pore size 220 nm) under vacuum. The nanowire network was transferred to a transparent substrate (glass or PET) by applying pressure to the back side of the membrane and forcing intimate contact with the substrate. The thin film was then annealed under forming gas (5% H2 and 95% Ar) at 160 °C for 60 min to improve junction contact before measurements.
Background substrate transmittances were subtracted from all the data. The characteristic transmittance and haze factors were acquired at a wavelength of 550 nm. The haze measurements were carried out using the D1003-13 standard. Four transmittance scans of a sample with different configurations were acquired for the haze calculations: T1, incident light; T2, total light transmitted by the specimen; T3, light scattered by the instrument; and T4, light scattered by the instrument and specimen. The haze factor of a specimen can be calculated by the equation haze = [(T4/T2) − (T3/T1)]%. A comparison of the performances of the transparent conductive film made of Cu NWs-FA/DT with the references is given in Extended Data Table 1.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Naboka, M. & Giordano, J. Copper Alloys: Preparation, Properties, and Applications (Nova Science, 2011).
Davis, J. R. (ed.) ASM Specialty Handbook: Copper and Copper Alloys (ASM International, 2001).
Kondo, K. et al. (eds) Copper Electrodeposition for Nanofabrication of Electronics Devices (Springer, 2014).
Laibinis, P. E. & Whitesides, G. M. Self-assembled monolayers of n-alkanethiolates on copper are barrier films that protect the metal against oxidation by air. J. Am. Chem. Soc. 114, 9022–9028 (1992).
Allam, N. K., Nazeer, A. A. & Ashour, E. A. A review of the effects of benzotriazole on the corrosion of copper and copper alloys in clean and polluted environments. J. Appl. Electrochem. 39, 961–969 (2009).
Jeong, S. et al. Controlling the thickness of the surface oxide layer on Cu nanoparticles for the fabrication of conductive structures by ink-jet printing. Adv. Funct. Mater. 18, 679–686 (2008).
Renner, F. U. et al. Initial corrosion observed on the atomic scale. Nature 439, 707–710 (2006).
Cui, C., Lim, A. T. O. & Huang, J. A cautionary note on graphene anti-corrosion coatings. Nat. Nanotechnol. 12, 834–835 (2017).
Finšgar, M. & Milošev, I. Inhibition of copper corrosion by 1,2,3-benzotriazole: a review. Corros. Sci. 52, 2737–2749 (2010).
Mihajlovic, M. B. P. & Antonijevic, M. M. Copper corrosion inhibitors. Period 2008–2014. A review. Int. J. Electrochem. Sci. 10, 1027–1053 (2015).
Niu, Z. et al. Ultrathin epitaxial Cu@Au core–shell nanowires for stable transparent conductors. J. Am. Chem. Soc. 139, 7348–7354 (2017).
Khan, M. H. et al. Atomically thin hexagonal boron nitride nanofilm for Cu protection: the importance of film perfection. Adv. Mater. 29, 1603937 (2017).
Dai, L. et al. Ultrastable atomic copper nanosheets for selective electrochemical reduction of carbon dioxide. Sci. Adv. 3, e1701069 (2017).
Mahvash, F. et al. Corrosion resistance of monolayer hexagonal boron nitride on copper. Sci. Rep. 7, 42139 (2017).
Lorenz, W. J. & Mansfeld, F. Determination of corrosion rates by electrochemical DC and AC methods. Corros. Sci. 21, 647–672 (1981).
Grillo, F., Tee, D. W., Francis, S. M., Fruchtl, H. A. & Richardson, N. V. Passivation of copper: benzotriazole films on Cu(111). J. Phys. Chem. C 118, 8667–8675 (2014).
Giessibl, F. J. Advances in atomic force microscopy. Rev. Mod. Phys. 75, 949–983 (2003).
Patera, L. L., Queck, F., Scheuerer, P. & Repp, J. Mapping orbital changes upon electron transfer with tunnelling microscopy on insulators. Nature 566, 245–248 (2019).
Barquín, M., González Garmendia, M. J., Larrínaga, L., Pinilla, E. & Torres, M. R. Complexes of copper(II) formate with 2-(phenylamino)pyridine and 2-(methylamino)pyridine: new copper formato paddle-wheel compounds. Inorg. Chim. Acta 359, 2424–2430 (2006).
Ye, B. H., Tong, M. L. & Chen, X. M. Metal–organic molecular architectures with 2, 2′-bipyridyl-like and carboxylate ligands. Coord. Chem. Rev. 249, 545–565 (2005).
Hafner, J. Ab-initio simulations of materials using VASP: density-functional theory and beyond. J. Comput. Chem. 29, 2044–2078 (2008).
Crapper, M. D., Riley, C. E., Woodruff, D. P., Puschmann, A. & Haase, J. Determination of the adsorption structure for formate on Cu(110) using SEXAFS and NEXAFS. Surf. Sci. 171, 1–12 (1986).
Stone, P., Poulston, S., Bennett, R. A., Price, N. J. & Bowker, M. An STM, TPD and XPS investigation of formic acid adsorption on the oxygen-precovered c(6×2) surface of Cu(110). Surf. Sci. 418, 71–83 (1998).
Chutia, A. et al. Adsorption of formate species on Cu(h,k,l) low index surfaces. Surf. Sci. 653, 45–54 (2016).
Kreikemeyer-Lorenzo, D. et al. Face-dependent bond lengths in molecular chemisorption: the formate species on Cu(111) and Cu(110). Phys. Rev. Lett. 107, 046102 (2011).
Jin, M. et al. Shape-controlled synthesis of copper nanocrystals in an aqueous solution with glucose as a reducing agent and hexadecylamine as a capping agent. Angew. Chem. Int. Ed. 50, 10560–10564 (2011).
Xu, L., Yang, Y., Hu, Z. W. & Yu, S. H. Comparison study on the stability of copper nanowires and their oxidation kinetics in gas and liquid. ACS Nano 10, 3823–3834 (2016).
Kolb, D. M. Reconstruction phenomena at metal-electrolyte interfaces. Prog. Surf. Sci. 51, 109–173 (1996).
Kim, Y.-G. et al. Surface reconstruction of pure-Cu single-crystal electrodes under CO-reduction potentials in alkaline solutions: a study by seriatim ECSTM-DEMS. J. Electroanal. Chem. 780, 290–295 (2016).
Gunathunge, C. M. et al. Spectroscopic observation of reversible surface reconstruction of copper electrodes under CO2 reduction. J. Phys. Chem. C 121, 12337–12344 (2017).
Li, X. et al. Large-area synthesis of high-quality and uniform graphene films on copper foils. Science 324, 1312–1314 (2009).
Li, S., Chen, Y., Huang, L. & Pan, D. Large-scale synthesis of well-dispersed copper nanowires in an electric pressure cooker and their application in transparent and conductive networks. Inorg. Chem. 53, 4440–4444 (2014).
Lee, Y., Choi, J., Lee, K. J., Stott, N. E. & Kim, D. Large-scale synthesis of copper nanoparticles by chemically controlled reduction for applications of inkjet-printed electronics. Nanotechnology 19, 415604 (2008).
Ahmad, Z. Principles of Corrosion Engineering and Corrosion Control (Elsevier, 2006).
Xu, H., Liu, Y., Chen, W., Du, R. G. & Lin, C. J. Corrosion behavior of reinforcing steel in simulated concrete pore solutions: a scanning micro-reference electrode study. Electrochim. Acta 54, 4067–4072 (2009).
Lin, C. J., Luo, J. L., Zhuo, X. D. & Tian, Z. W. Scanning microelectrode studies of early pitting corrosion of 18/8 stainless steel. Corrosion 54, 265–270 (1998).
Biesinger, M. C., Lau, L. W. M., Gerson, A. R. & Smart, R. S. C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V, Cu and Zn. Appl. Surf. Sci. 257, 887–898 (2010).
Hapala, P. et al. Mechanism of high-resolution STM/AFM imaging with functionalized tips. Phys. Rev. B 90, 085421 (2014).
Hapala, P., Temirov, R., Tautz, F. S. & Jelinek, P. Origin of high-resolution IETS-STM images of organic molecules with functionalized tips. Phys. Rev. Lett. 113, 226101 (2014).
Zhao, L. et al. A force field for dynamic Cu-BTC metal–organic framework. J. Mol. Model. 17, 227–234 (2011).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Kresse, G. & Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Crapper, M. D., Riley, C. E. & Woodruff, D. P. The structure of formate on Cu(100) and Cu(110) surfaces. Surf. Sci. 184, 121–136 (1987).
Leibsle, F. M., Haq, S., Frederick, B. G., Bowker, M. & Richardson, N. V. Molecularly induced step faceting on Cu(110) surfaces. Surf. Sci. 343, L1175–L1181 (1995).
Poulston, S., Bennett, R. A., Jones, A. H. & Bowker, M. STM study of formic acid adsorption on Cu(110). Phys. Rev. B 55, 12888–12891 (1997).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Jing, J. L. & Yan, M. L. Statistical entropy of a stationary dilaton black hole from the Cardy formula. Phys. Rev. D 63, 024003 (2000).
Haynes, W. M. CRC Handbook of Chemistry and Physics (CRC, 2014).
Gokhale, A. A., Kandoi, S., Greeley, J. P., Mavrikakis, M. & Dumesic, J. A. Molecular-level descriptions of surface chemistry in kinetic models using density functional theory. Chem. Eng. Sci. 59, 4679–4691 (2004).
Richard, F. & Bader, R. Atoms in Molecules (A Quantum Theory) (Clarendon Press, 1990).
Grundner, S. et al. Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol. Nat. Commun. 6, 7546 (2015).
Li, G. et al. Stability and reactivity of copper oxo-clusters in ZSM-5 zeolite for selective methane oxidation to methanol. J. Catal. 338, 305–312 (2016).
Rathmell, A. R., Bergin, S. M., Hua, Y. L., Li, Z. Y. & Wiley, B. J. The growth mechanism of copper nanowires and their properties in flexible, transparent conducting films. Adv. Mater. 22, 3558–3563 (2010).
Guo, H. et al. Copper nanowires as fully transparent conductive electrodes. Sci. Rep. 3, 2323 (2013).
Ahn, Y., Jeong, Y., Lee, D. & Lee, Y. Copper nanowire–graphene core–shell nanostructure for highly stable transparent conducting electrodes. ACS Nano 9, 3125–3133 (2015).
Song, J., Li, J., Xu, J. & Zeng, H. Superstable transparent conductive Cu@Cu4Ni nanowire elastomer composites against oxidation, bending, stretching, and twisting for flexible and stretchable optoelectronics. Nano Lett. 14, 6298–6305 (2014).
Rathmell, A. R., Nguyen, M., Chi, M. & Wiley, B. J. Synthesis of oxidation-resistant cupronickel nanowires for transparent conducting nanowire networks. Nano Lett. 12, 3193–3199 (2012).
Cheng, Y., Wang, S. L., Wang, R. R., Sun, J. & Gao, L. Copper nanowire based transparent conductive films with high stability and superior stretchability. J. Mater. Chem. C 2, 5309–5316 (2014).
Mayousse, C., Celle, C., Carella, A. & Simonato, J. P. Synthesis and purification of long copper nanowires. Application to high performance flexible transparent electrodes with and without PEDOT:PSS. Nano Res. 7, 315–324 (2014).
Stewart, I. E., Ye, S. R., Chen, Z. F., Flowers, P. F. & Wiley, B. J. Synthesis of Cu–Ag, Cu–Au, and Cu–Pt core–shell nanowires and their use in transparent conducting films. Chem. Mater. 27, 7788–7794 (2015).
Aliprandi, A. et al. Hybrid copper-nanowire–reduced-graphene-oxide coatings: a “green solution” toward highly transparent, highly conductive, and flexible electrodes for (opto)electronics. Adv. Mater. 29, 1703225 (2017).
Chen, Z., Ye, S., Stewart, I. E. & Wiley, B. J. Copper nanowire networks with transparent oxide shells that prevent oxidation without reducing transmittance. ACS Nano 8, 9673–9679 (2014).
Xiong, W. et al. Highly conductive, air-stable silver nanowire@Iongel composite films toward flexible transparent electrodes. Adv. Mater. 28, 7167–7172 (2016).
Ge, Y. J. et al. Direct room temperature welding and chemical protection of silver nanowire thin films for high performance transparent conductors. J. Am. Chem. Soc. 140, 193–199 (2018).
Deng, Y. L., Handoko, A. D., Du, Y. H., Xi, S. B. & Yeo, B. S. In situ Raman spectroscopy of copper and copper oxide surfaces during electrochemical oxygen evolution reaction: identification of CuIII oxides as catalytically active species. ACS Catal. 6, 2473–2481 (2016).
Decremps, F., Pellicer-Porres, J., Saitta, A. M., Chervin, J. C. & Polian, A. High-pressure Raman spectroscopy study of wurtzite ZnO. Phys. Rev. B 65, 092101 (2002).
Honesty, N. R. & Gewirth, A. A. Shell-isolated nanoparticle enhanced Raman spectroscopy (SHINERS) investigation of benzotriazole film formation on Cu(100), Cu(111), and Cu(poly). J. Raman Spectrosc. 43, 46–50 (2012).
Luo, W., Xu, Y. M., Wang, Q. M., Shi, P. Z. & Yan, M. Effect of grain size on corrosion of nanocrystalline copper in NaOH solution. Corros. Sci. 52, 3509–3513 (2010).
Quraishi M. A. Electrochemical and theoretical investigation of triazole derivatives on corrosion inhibition behavior of copper in hydrochloric acid medium. Corros. Sci. 70, 161–169 (2013).
Bodappa, N. et al. Early stages of electrochemical oxidation of Cu(111) and polycrystalline Cu surfaces revealed by in situ Raman spectroscopy. J. Am. Chem. Soc. 141, 12192–12196 (2019).
Li, J. F. et al. Shell-isolated nanoparticle-enhanced Raman spectroscopy. Nature 464, 392–395 (2010).
Poulston, S., Parlett, P. M., Stone, P. & Bowker, M. Surface oxidation and reduction of CuO and Cu2O studied using XPS and XAES. Surf. Interface Anal. 24, 811–820 (1996).
Speckmann, H. D., Haupt, S. & Strehblow, H. H. A quantitative surface analytical study of electrochemically-formed copper oxides by XPS and X-ray-induced Auger-spectroscopy. Surf. Interface Anal. 11, 148–155 (1988).
Cano, E., Torres, C. L. & Bastidas, J. M. An XPS study of copper corrosion originated by formic acid vapour at 40% and 80% relative humidity. Mater. Corros. 52, 667–676 (2001).
Inui, T., Koga, H., Nogi, M., Komoda, N. & Suganuma, K. A miniaturized flexible antenna printed on a high dielectric constant nanopaper composite. Adv. Mater. 27, 1112–1116 (2015).
Acknowledgements
We acknowledge financial support from the National Key R&D Program of China (2017YFA0207302, 2017YFA0207303, 2016YFA0300901 and 2017YFA0205003), the National Natural Science Foundation of China (21890752, 21731005, 21721001, 11634001, 21725302, 11888101, 21905237 and 91845102), the China Postdoctoral Science Foundation (2017M612131), the Beijing Municipal Science & Technology Commission (Z191100007219005) and the Tencent Foundation through XPLORER PRIZE. Part of this research used Beamline 02B and Beamline 03U of the Shanghai Synchron Radiation Facility, which is supported by ME2 project under contract no. 11227902 from the National Natural Science Foundation of China. We thank H. Häkkinen for discussions, and Y. Cao and W. Song for providing the Cu–graphene sample. We also thank P. Cheng from Analytical Instrumentation Center, SPST, Shanghai Tech University for help with the XPS measurement.
Author information
Authors and Affiliations
Contributions
N.Z. conceived and supervised the research project. J.P. synthesized and characterized the samples and investigated their anti-corrosion performance. G.F. supervised the DFT calculations and analysed the computational results together with B.C., who conducted the calculations. Y.J. supervised the STM and AFM measurements, which were carried out by Z.W., J.G., S.Y. and D.C.; Q. Zhang and L.G. contributed to the HRTEM characterizations of Cu-FA samples. S. Hao, A.F., H.X., S. Hong, and B.W. contributed to the sample preparations and characterizations. C.-J.L. supervised the SRET measurements. L.-S.Z. and Z.S. contributed to the TPD-MS measurements. Q. Zhou and Z.L. contributed to the XPS measurements. N.Z., G.F., Y.J., J.P., B.C., Z.W. and J.G. wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks C. Richard Catlow and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Anti-corrosion properties of Cu-FA.
Cu-FA shows outstanding anti-corrosion properties while maintaining the excellent thermal and electrical conductivities of Cu, as revealed by qualitative and quantitative evaluations of the anti-corrosion performances of Cu foils before and after formate treatment. a, Optical and SEM images (left) and Raman spectra (right) of brass, bronze, Cu-G and Cu-BTA before and after corrosion in 0.1 M NaOH at 25 °C for 8 h. The Raman bands in the spectral ranges 170–230 cm−1, and 270–320 cm−1 and 590–680 cm−1 are attributed to Cu–O vibrations from Cu2O and CuO species68, respectively. The weak Raman bands in the range 564–600 cm−1 come from ZnO species69. The Raman band at ~535 cm−1 is assigned to the triazole ring bending mode70. b, Optical image (top), Raman spectra (middle) and XRD patterns (bottom) of the Cu foil and Cu-FA before and after corrosion in 0.1 M NaOH for 12 h. Raman bands centred at 298 cm−1 and 628 cm−1 come from CuO species. The XRD pattern of the Cu foil after corrosion reveals the formation of CuO. By contrast, Cu-FA remains almost the same after the 12-h corrosion as before the test. c, d, Electric (c) and thermal (d) conductivities of Cu, Cu-FA (prepared by Method I), brass and bronze foils before and after corrosion in 0.1 M NaOH at room temperature for 12 h. e, Microphotograph and corresponding Raman spectrum showing the corrosion of the Cu foils before treatment in a sodium formate–H2O mixture at 160 °C for 24 h. In the corrosion test, the foils were immersed in 0.1 M NaOH at 25 °C for 24 h. The two groups of Raman bands observed at (146, 217, 417, 532) cm−1 and (307, 628) cm−1 are assigned to Cu2O and CuO species, respectively68. f, Microphotograph and corresponding Raman spectra of Cu-FA before and after corrosion in 0.1 M NaOH at 25 °C for 24 h. (g) Cyclic voltammetry (CV) curves of bare Cu and Cu-FA in 0.1 M NaOH. Two anodic current peaks and two cathodic current peaks are observed for bare Cu, which can be assigned to the two Cu redox reactions. Whereas the anodic peaks at cell potentials of −0.30 V and −0.10 V are due to the forward reactions, the cathodic peaks at −0.40 V and −0.75 V are attributed to the reverse reactions. However, no oxidation peaks were observed for Cu-FA, indicating the substantial suppression of Cu oxidation. h, Tafel plots of bare Cu and Cu-FA in 0.1 M NaOH. The Tafel plot of bare Cu is in good accordance with the reference71. Although there is slight change in the peak potential, the corrosion current density of Cu-FA is 20 times lower than that of bare Cu. The slight shift in the Tafel plot of Cu-FA suggests that the transfer of oxygen from the bulk solution to the cathodic sites of Cu is inhibited by the formate treatment72. The polarization parameters of bare Cu and Cu-FA in 0.1 M NaOH are: Ecorr = −222 mV, Jcorr = 6.71 μA cm−2, corrosion rate 78.2 μm yr−1 (bare Cu); Ecorr = −213 mV, Jcorr = 0.33 μA cm−2, corrosion rate 3.89 μm yr−1 (Cu-FA); anti-corrosion factor 20.1. The anti-corrosion factor is defined as the ratio of the corrosion rate of the bare Cu foil to that of the modified Cu in 0.1 NaOH. i, Raman spectra of bare Cu and Cu-FA after electrochemical tests. The Raman bands at (149, 217) cm−1 and 636 cm−1 come from Cu2O and CuO species, respectively68. j, Nyquist impedance plots of bare Cu and Cu-FA at 0.1 V versus Ag/AgCl. At high frequency, the capacitive impedance of the electrode–electrolyte interface becomes more effective at shunting the charge-transfer resistance. Therefore, the charge-transfer resistance calculated from the impedance difference at lower and higher frequencies is used to qualitatively evaluate the corrosion rate. Compared with the bare Cu, a 14-fold increase in the charge-transfer resistance is observed on Cu-FA, indicating that the corrosion is indeed strongly inhibited by the FA modification. Z′ and Z″ are the real and imaginary parts of the impedance, respectively.
Extended Data Fig. 2 Comparison of anti-corrosion performance of Cu-FA with Cu foils with other corrosion inhibitors.
a, Electrochemical characterizations of Cu-FA foil and Cu foils treated with DT and BTA corrosion inhibitors. Whereas all measurements were carried out in 0.1 M NaOH, the Nyquist impedance and Bode plots were recorded at 0.1 V versus Ag/AgCl. The polarization parameters of Cu foils with different surface treatments in 0.1 M NaOH are: Ecorr = −212 mV, Jcorr = 5.12 μA cm−2, corrosion rate 59.7 μm yr−1, anti-corrosion factor 1.31 (Cu-BTA); Ecorr = −210 mV, Jcorr = 4.33 μA cm−2, corrosion rate 50.4 μm yr−1, anti-corrosion factor 1.55 (Cu-DT). For comparison, the anti-corrosion factor of Cu-FA in 0.1 M NaOH is 20.1. b, Tafel and Nyquist plots, SEM images and Raman spectra of the samples before and after the electrochemical measurements. The Nyquist plots were measured at 0.1 V versus Ag/AgCl in 1 M NaOH. The Raman bands at 297 cm−1 and 623 cm−1 come from CuO species68. Owing to the concentration difference, the Tafel plot of Cu-FA in 1 NaOH displays a minimum that is different from that in 0.1 M NaOH. The polarization parameters of Cu foils before and after the formate treatment in 1.0 M NaOH are: Ecorr = −281 mV, Jcorr = 59.7 μA cm−2, corrosion rate 695.8 μm yr−1 (bare Cu); Ecorr = −311 mV, Jcorr = 5.10 μA cm−2, corrosion rate 59.4 μm yr−1 (Cu-FA), anti-corrosion factor 11.7. The anti-corrosion factor is defined as the ratio of the corrosion rate bare Cu foil to that of modified Cu in 1 NaOH. c, SEM images of bare Cu and Cu-FA (prepared by Method II) foils after exposure to 30% H2O2 for 0, 5 and 15 min. d, Optical images of Cu-FA (prepared by Method II) and untreated Cu foils before and after heating at 160 °C in air for 1 h and corresponding Raman spectra after thermal treatment. e, Localized corrosion of Cu-FA monitored by SRET/STM. The three-dimensional corrosion current maps were recorded on scratched Cu-FA and bare Cu foils after immersion in 0.1 M NaOH for 5, 30 and 90 min, and SEM images before and after the SRET/STM test were taken to identify the oxidized area.
Extended Data Fig. 3 Effect of treatment time and crystallinity on the anti-corrosion performance of Cu.
a, Photographs and microphotographs of Cu-FA foils after corrosion in 0.1 M NaOH for 24 h as a function of treatment time. The foils were obtained with the formate treatment at 160 °C for different periods. b, XRD spectra of different Cu foil samples treated hydrothermally in the presence of formate in different conditions. Sample 1: Cu-FA foil (25 μm; Alfa Asear) treated at 160 °C for 0–12 h (Method II). Sample 2: Cu foil (25 μm; Alfa Asear) treated at 200 °C for 2–24 h (Method I). Sample 3: Cu foil (10 μm; Aladdin) treated at 200 °C for 0–4 h (Method I). c, Cross-sectional TEM and high-resolution STEM images of Cu-FA (I–II) and bare Cu foils (III–IV), using samples prepared by the focused-ion-beam technique. d, Cross-sectional TEM and high-resolution TEM (HRTEM) images of Cu-FA (1–3) and bare Cu foils (4–6), using samples prepared by microtoming.
Extended Data Fig. 4 Identification of the surface coordinating species on Cu-FA.
a, STM images of Cu-FA foils (prepared by Method II) and untreated Cu foils after annealing for 4 h in UHV at 150 °C or 300 °C. A step height of 0.258 nm, a diatomic step height of Cu(110), was readily observed after annealing at 150 °C, although the high-resolution STM images were obtained only after annealing at 300 °C. In comparison, no ordered structure was observed on untreated Cu foils with different annealing conditions. b, STM images showing the successful formation of the c(6 × 2) superstructure on single-crystal Cu(110) treated with sodium formate solution followed by annealing at 150 °C, which is similar to the temperature used in the formate treatment to create effective passivation. The presence of dark depressions (highlighted by yellow arrows) may arise from interstitial O2− species of the hydrated c(6 × 2) surface structure. c, Structure models of the paddle-wheel dinuclear Cu(ii) formate complex and the Cu(110) surface co-passivated by [Cu(μ-HCOO)(OH)2]2 and O2−. d, FTIR, Raman and TPD-MS spectra of the Cu-FA foils (prepared by Method II) after annealing under the same conditions as those used for STM imaging and re-exposure to air. The inset in the Raman panel is the optical photograph of the annealed sample in air. The presence of a broad infrared absorption band at 3,378 cm-1 clearly suggests the presence of abundant -OH groups on the surface. The Raman spectrum was obtained by using Au@SiO2 SHINERS particles to enhance the Raman signals (524 cm−1 for Cu-O, 1,404 and 2,920 cm−1 for the C–H vibration (νC–H) on formate)73,74. Both control Raman spectra of the SHINERS particles and Cu-FA foils are given for comparison. The TPD-MS profiles clearly show the release of H2O and HCOO− species from Cu-FA in a wide range of temperatures. The inset in the TPD-MS spectra shows the relative ionization intensities of the detected species. e, Tafel curves of Cu-FA before and after annealing. f, Linear sweep voltammetry of Cu-FA after annealing, obtained with a scan rate of 2 mV s−1 from −1.2 V to 0 V (versus Ag/AgCl) in 0.1 M NaHCO3 solution (pH 8). The Cu(ii) and Cu(i) species were detected with redox potentials in good accordance with reported values52. g, XPS spectra of bare Cu and Cu-FA foils after annealing under the same conditions as those used for STM/AFM characterizations. Once exposed to air, the bare Cu foil displays obvious peaks (935.1, 940.8 and 944.1 eV) corresponding to the oxidized Cu species. In comparison, the Cu 2p XPS (full-width at half-maximum, FWHM = 0.9 eV) and Cu LMM Auger spectra of annealed samples of bare Cu (without air exposure) and Cu-FA foils are almost identical and display peaks of metallic Cu reported in the literature75,76. These data clearly suggest the presence of a non-detectable amount of oxidized Cu species on annealed Cu-FA. Whereas the presence of OH− is clearly observed in the O 1s XPS spectrum of the unannealed Cu-FA foil, the annealed Cu-FA displays O 1s XPS signals from O from the formate (532.3 eV, FWHM = 1.7 eV) and O2− on Cu (530.4 eV, FWHM = 1.2 eV) in a ratio close to 1. No obvious OH− signal is identified on the annealed sample. In comparison, the bare Cu foil after air exposure displays a major O 1s XPS signal corresponding to OH−. The annealed Cu foil shows O 1s XPS signals at 530.9 (FWHM = 1.2 eV), 531.9 (FWHM = 1.7 eV) and 533.4 (FWHM = 1.3 eV), corresponding to O2−, OH− and O from the carbonate, respectively77. The C 1s spectra of both annealed Cu and Cu-FA foils show the dominant presence of carbon contamination.
Extended Data Fig. 5 The importance of Cu(110) for the anti-corrosion properties.
a, STM topographies of the single-crystal Cu(110)-c(6 × 2) sample after exposure to air and then annealing at 120 °C. The zoom-in STM image shows distortion and darker depressions in the Cu(110)-c(6 × 2) superstructure, suggesting the occurrence of a hydration process during air exposure. b, STM image of the single-crystal Cu(110)-c(6 × 2) sample after air exposure followed by annealing at 300 °C. The regeneration of the dehydrated c(6 × 2) structure without dark depressions was confirmed. c, Structure models showing the adsorption of O2 and Cl− on clean Cu(110) (I, II) and FA-modified Cu(110) (III, IV). O2 is easily dissociated on clean Cu(110) to form adsorbed O species. The Bader charge of Cu atoms on the modified Cu(110) and reference systems was as follows: (1) modified Cu(110): surface Cu +0.97 to +1.0, subsurface Cu +0.34 to +0.53, bulk Cu 0 to +0.14; (2) reference systems: bulk CuO +0.99, bulk Cu2O +0.57, Cu(110) +0.01 to −0.02. d, XRD patterns of Cu(100), Cu(111) and Cu(110) single crystals. e, XRD patterns of scratched Cu(111) single crystal treated with formate at 160 °C for 0–60 h. If the surface was not scratched, no change on the XRD pattern was detected even for treatment time of up to 60 h. f, Micrographs and Raman spectra of Cu(110), Cu(100) and Cu(111) single-crystal samples treated by an aqueous solution of formate at 100 °C for 1 h, 10 h and 10 h, respectively. The two groups of Raman bands at (146, 217, 416, 535) cm−1 and 635 cm−1 are attributed to the Cu2O and CuO species, respectively68.
Extended Data Fig. 6 Enhanced anti-corrosion performance in harsh conditions by introducing alkanethiol.
a, Structure model of Cu-FA with alkanethiol bound to the step sites, and experimental evidence of the successful introduction of 1-DT ligands. Whereas the change of contact angle of the water droplets suggests the change of surface hydrophobicity, TPD-MS confirms the presence of formate and DT on Cu-FA/DT. The Cu-FA foils were prepared by Method III. b, Electrochemical measurements (CV, Tafel, Nyquist impedance and Bode plots) confirming the enhanced anti-corrosion properties of Cu-FA after the DT treatment. All measurements were carried out in 0.1 M NaOH solution. The Nyquist impedance and Bode plots were measured at 0.1 V versus Ag/AgCl. The polarization parameters of Cu-FA/DT foils in 0.1 M NaOH and 1 M NaOH are: Ecorr = −258 mV, Jcorr = 0.067 μA cm−2, corrosion rate 0.755 μm yr−1, anti-corrosion enhancement 103.6 (0.1 M NaOH); Ecorr = −331 mV, Jcorr = 0.38 μA cm−2, corrosion rate 4.40 μm yr−1, anti-corrosion factor 158.1 (1 M NaOH). The anti-corrosion factor is defined as the ratio of the corrosion rate of the bare Cu foil to that of the modified Cu in the same NaOH solutions. c, Comparison of optical images and Raman spectra for the evaluation of the enhanced anti-corrosion of Cu-FA (prepared by Method III) against salt spray after introducing DT. Before the measurements, Cu, Cu-FA and Cu-FA/DT foil samples were subjected to a 5% NaCl salt spray test at 47 °C for 24 h. The Raman bands around (149, 217) cm−1 and 640 cm−1 are attributed to Cu2O and CuO species, respectively68. d, Comparison of optical imaging data that reveals the enhanced anti-corrosion of Cu-FA/DT against Na2S. Before recording the images, both Cu-FA and Cu-FA/DT foils were exposed to Na2S solutions with concentrations ranging from 0.1 mM to 1 M for 1 h. Compared to Cu-FA, Cu-FA/DT could survive and keep its copper colour in a much higher concentration of Na2S. e, SEM images and Raman spectra of Cu-FA and Cu-FA/DT after treatment in 10 mM Na2S for 1 h and 5 h, respectively. The Raman bands around 296 cm−1 and 624 cm−1 indicate the formation of oxidized and sulfurized Cu species. f, SEM images of Cu, Cu-FA and Cu-FA/DT foils after seawater inundation at room temperature for 30 days. g, Photographs showing enhanced anti-corrosion of Cu-FA/DT in H2O2. The photographs of the Cu-FA/DT, Cu-FA and Cu foils were taken after they were immersed in 30% H2O2 for different intervals.
Extended Data Fig. 7 Outstanding anti-corrosion performances regardless of the shape and size of Cu materials.
a, SEM images of Cu and Cu-FA (prepared by Method II) wires before and after treatment in 0.1 M NaOH at 60 °C for 60 h and in 3.5 wt% NaCl at 60 °C for 24 h (top), temperature-dependent resistances of Cu and Cu-FA wires (length 10 cm, diameter 1 mm) after heating in air for 24 h (bottom left) and resistance changes of Cu and Cu-FA (prepared by Method II) wires (length 10 cm, diameter 1 mm) after ageing in 0.1 M NaOH at 60 °C for different time periods (bottom right). The data were averaged from three independent measurements. Error bars reflect the standard errors. b, SEM images of the untreated Cu wire and the Cu-FA wire before and after heating at 160 °C in air for 24 h. c, Microphotographs of Cu, Cu-DT, Cu-FA (prepared by Method III) and Cu-FA/DT meshes after a 96-h salt spray test, and their corresponding Raman spectra and relative electric conductivities before (grey) and after (red) the salt spray test. d, Optical photographs of Cu-FA/DT and Cu-FA meshes (prepared by Method III) after immersion in Na2S solutions of different concentrations for 5 h, and microphotographs of Cu–FA/DT and Cu-FA meshes after 6 h of ageing in 10 mM Na2S. e, Robust anti-corrosion performance of Cu-FA/DT tubes under wet mechanical conditions; photographs of the outer and inner walls of Cu-FA/DT and bare Cu tubes (inner diameter 1.6 cm) after the wet mechanical test with flowing 3.5% NaCl solutions (flow rate of up to 1,400 l h−1, 10–30 °C) for 96 h and 12 h, respectively, and their corresponding Raman spectra. f, Robust anti-corrosion performance of Cu-FA/DT foils under wet mechanical conditions; photographs of the wet mechanical test set-up, with salty water (3.5% NaCl, 1% Na2CO3, 1% Na2SO4 and 0.1% NaOH) flowing through the foils, comparison photographs of Cu-FA/DT, bare Cu and patinated Cu foils before and after the wet mechanical tests (72, 12 and 12 h, respectively) and their corresponding Raman spectra after the tests. The polarization parameters of different Cu foils in 0.1 M NaOH solution were measured as follows: Ecorr = −222 mV, Jcorr = 6.71 μA cm−2, corrosion rate 78.2 μm yr−1 (bare Cu); Ecorr = −212 mV, Jcorr = 4.65 μA cm−2, corrosion rate 54.2 μm yr−1, anti-corrosion enhancement 1.44 (patinated Cu); Ecorr = −258 mV, Jcorr = 0.067 μA cm−2, corrosion rate 0.755 μm yr−1, anti-corrosion enhancement 103.6 (Cu-FA/DT). The anti-corrosion enhancement is defined as the ratio of the corrosion rate of the bare Cu foil to that of modified Cu. The patinated Cu foil was prepared by exposing bare Cu foils to 3.5% Na2CO3 salt spray for 60 h.
Extended Data Fig. 8 Application of the anti-corrosion technique to Cu nanomaterials.
a, SEM images of fresh Cu NWs and of untreated Cu NWs exposed to air for 5 days, showing the easy oxidation of untreated Cu NWs. b, XRD patterns of Cu NWs (prepared by Method III), Cu NWs-FA and Cu NWs-FA/DT after a 12-h corrosion test in 1 M NaOH (left), photographs of their water suspensions before and after being stored at room temperature in air for 90 days (top right) and their Raman spectra after storage in air for 5 and 30 days (bottom right). c, TEM and HRTEM images of Cu NWs and Cu NWs-FA. The insets display the corresponding selected-area electron diffraction patterns. d, TEM and HRTEM images of Cu NWs, Cu NWs-FA and Cu NWs-FA/DT before and after 24-h corrosion tests in 1 M NaOH. Typical lattice fringes are labelled in the HRTEM images. e, SEM images of Cu NWs, Cu NWs-FA and Cu NWs-FA/DT after heating at 80 °C for 24 h. f, Performance of passivated Cu NWs in transparent electrodes. The transmittance is shown as a function of the sheet resistance of transparent electrodes made from Cu NWs with different surface modifications. The inset shows a photograph of a transparent electrode with Cu NWs on PET. Together with the resistance change of the Cu NWs-FA/DT film during a bending test with sputtered indium tin oxide (ITO), the resistance changes of transparent electrodes made from untreated Cu NWs, Cu NWs-FA, Cu NWs/DT and Cu NWs-FA/DT after heating at 80 °C in air for 24 h are also provided for comparison. The data were averaged from three independent measurements. Error bars represent the standard errors. g, SEM images of Cu NWs, Cu NWs-FA and Cu NWs-FA/DT before and after a 24-h salt spray test. h, Change of the square resistance of Cu NPs and Cu NPs-FA (prepared by Method II) films after storage at ambient conditions for different periods. The measurements were carried out on films made from Cu NPs and Cu NPs-FA with a thickness of 100 μm. The insets show photographs of Cu NPs and Cu NPs-FA powders after a 14-day exposure to air at room temperature.
Extended Data Fig. 9 Application of anti-oxidation Cu materials to radio-frequency identification, and room-temperature electrochemical technique for preparing anti-corrosive Cu materials.
a, Photograph of kilogram-scale anti-corrosion Cu paste. b, Photograph of radio-frequency identification (RFID) tag antenna. This antenna was designed for ultrahigh-frequency RFID on a cloth substrate (the copper paste was printed on the cloth substrate by screen printing). The working frequency is 915 MHz, and its performance is equivalent to that of similar commercial aluminium-etching antennas. The recognition distance can reach 20.3 m. c, Reflection coefficient S11 of the antenna shown in b, measured by Vector network analyser ZNB8. The reflection coefficient S11 is the most important parameter of the antenna; a smaller S11 implies lower energy loss of the tag antenna in the process of receiving the signal from the card reader, that is, higher energy utilization. These data verify that the antenna works at 915 MHz and also show that the S11 value of the antenna at this frequency can reach −41.5 dB (a very low value), which is equivalent to S11 values reported in the literature78. This suggests that our copper paste can also be used to create high-efficiency and high-performance antennas. d, Stability of the electrical conductivity of our Cu-FA/DT-paste-based RFID tag antenna compared to that of a paste of untreated Cu. For copper-paste-based RFID films, the square resistance was acquired and S = 1/(sheet resistance × thickness of the foils or films). e, CV curve of Cu foil immersed in 2% HCOONa (pH 8–10) recorded at a scan rate of 10 mV s−1, and chronoamperometric curve with reduction potential −0.7 V (versus SCE) applied after an anodic sweep from −0.80 V to 0.20 V (versus SCE; 10 mV s−1). f, Photographs of Cu-FA(EC) and Cu foils before and after a 12-h corrosion test in 0.1 M NaOH. g, SEM images and Raman spectra of Cu-FA(EC) and bare Cu foils after the corrosion test in NaOH. h, Microphotographs and Raman spectra of Cu-FA(EC)/DT, Cu-FA(EC) and Cu foils after immersion in 50 mM Na2S solution for 10 min. i, Schematic illustration of the roll-to-roll setup used to produce Cu-FA(EC). RE, reference electrode; CE, counter electrode; WE, working electrode. An oxidation potential of 0.1 V versus SCE was applied for the generation of surface Cu(ii) when the Cu foil was moving from left to right, and an oxidation potential of −0.7 V versus SCE was used when the foil was moving back from right to left for the surface reconstruction.
Rights and permissions
About this article

Cite this article
Peng, J., Chen, B., Wang, Z. et al. Surface coordination layer passivates oxidation of copper. Nature 586, 390–394 (2020). https://doi.org/10.1038/s41586-020-2783-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2783-x
This article is cited by
-
Passivation and corrosion of Al current collectors in lithium-ion batteries
npj Materials Degradation (2024)
-
Copper Bonding Technology in Heterogeneous Integration
Electronic Materials Letters (2024)
-
A critical review of operating stability issues in electrochemical CO2 reduction
Science China Materials (2024)
-
Significantly improved electrochemical performance of the commercial lithium titanate (Li4Ti5O12) achieved by using a novel current collector of cuprous iodide-modified copper foil
Bulletin of Materials Science (2024)
-
Ag and Sn capping layers for stabilizing Cu solar cell contacts against oxidation in air: effects of method and solution of deposition
Journal of Materials Science: Materials in Electronics (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
