
Abstract
The detection of oral bacteria in faecal samples has been associated with inflammation and intestinal diseases. The increased relative abundance of oral bacteria in faeces has two competing explanations: either oral bacteria invade the gut ecosystem and expand (the ‘expansion’ hypothesis), or oral bacteria transit through the gut and their relative increase marks the depletion of other gut bacteria (the ‘marker’ hypothesis). Here we collected oral and faecal samples from mouse models of gut dysbiosis (antibiotic treatment and DSS-induced colitis) and used 16S ribosomal RNA sequencing to determine the abundance dynamics of oral bacteria. We found that the relative, but not absolute, abundance of oral bacteria increases, reflecting the ‘marker’ hypothesis. Faecal microbiome datasets from diverse patient cohorts, including healthy individuals and patients with allogeneic haematopoietic cell transplantation or inflammatory bowel disease, consistently support the ‘marker’ hypothesis and explain associations between oral bacterial abundance and patient outcomes consistent with depleted gut microbiota. By distinguishing between the two hypotheses, our study guides the interpretation of microbiome compositional data and could potentially identify cases where therapies are needed to rebuild the resident microbiome rather than protect against invading oral bacteria.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


Data availability
A comprehensive list of the microbiome datasets used in this study, along with an indication of whether they contain quantitative data, is provided in Supplementary Table 9. The raw 16S sequences of mouse faecal and oral samples collected in this study have been uploaded to the Sequence Read Archive (SRA) with BioProject accession number PRJNA873058. All other microbiome datasets are publicly available. All processed data supporting the findings of this study are available within the Article and its supplementary materials.
Code availability
Customized Python codes for reproducing the figures and tables are available on GitHub (https://github.com/liaochen1988/Oral_bacterial_manuscript_revision).
References
The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012).
Segata, N. et al. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 13, R42 (2012).
Atarashi, K. et al. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science 358, 359–365 (2017).
Li, B. et al. Oral bacteria colonize and compete with gut microbiota in gnotobiotic mice. Int. J. Oral Sci. 11, 10 (2019).
Kitamoto, S. et al. The intermucosal connection between the mouth and gut in commensal pathobiont-driven colitis. Cell 182, 447–462.e14 (2020).
Kitamoto, S., Nagao-Kitamoto, H., Hein, R., Schmidt, T. & Kamada, N. The bacterial connection between the oral cavity and the gut diseases. J. Dent. Res. 99, 1021–1029 (2020).
Jin, S., Wetzel, D. & Schirmer, M. Deciphering mechanisms and implications of bacterial translocation in human health and disease. Curr. Opin. Microbiol. 67, 102147 (2022).
Abed, J. et al. Colon cancer-associated Fusobacterium nucleatum may originate from the oral cavity and reach colon tumors via the circulatory system. Front. Cell. Infect. Microbiol. 10, 400 (2020).
Schmidt, T. S. et al. Extensive transmission of microbes along the gastrointestinal tract. eLife 8, e42693 (2019).
Rashidi, A., Ebadi, M., Weisdorf, D. J., Costalonga, M. & Staley, C. No evidence for colonization of oral bacteria in the distal gut in healthy adults. Proc. Natl Acad. Sci. USA 118, e2114152118 (2021).
Gevers, D. et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392 (2014).
Schirmer, M. et al. Compositional and temporal changes in the gut microbiome of pediatric ulcerative colitis patients are linked to disease course. Cell Host Microbe 24, 600–610.e4 (2018).
Priya, S. et al. Identification of shared and disease-specific host gene–microbiome associations across human diseases using multi-omic integration. Nat. Microbiol. 7, 780–795 (2022).
Kostic, A. D. et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14, 207–215 (2013).
Qin, N. et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64 (2014).
Reikvam, D. H. et al. Depletion of murine intestinal microbiota: effects on gut mucosa and epithelial gene expression. PLoS ONE 6, e17996 (2011).
Taguer, M. et al. Changes in gut bacterial translation occur before symptom onset and dysbiosis in dextran sodium sulfate-induced murine colitis. mSystems 6, e0050721 (2021).
Shenhav, L. et al. FEAST: fast expectation-maximization for microbial source tracking. Nat. Methods 16, 627–632 (2019).
Imai, J. et al. A potential pathogenic association between periodontal disease and Crohn’s disease. JCI Insight 6, e148543 (2021).
Liao, C. et al. Compilation of longitudinal microbiota data and hospitalome from hematopoietic cell transplantation patients. Sci. Data 8, 71 (2021).
Bekker, V. et al. Dynamics of the gut microbiota in children receiving selective or total gut decontamination treatment during hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 25, 1164–1171 (2019).
Yan, J. et al. A compilation of fecal microbiome shotgun metagenomics from hematopoietic cell transplantation patients. Sci. Data 9, 219 (2022).
Read, E., Curtis, M. A. & Neves, J. F. The role of oral bacteria in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 18, 731–742 (2021).
Vandeputte, D. et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511 (2017).
Contijoch, E. J. et al. Gut microbiota density influences host physiology and is shaped by host and microbial factors. eLife 8, e40553 (2019).
Britton, G. J. et al. Defined microbiota transplant restores Th17/RORγt+ regulatory T cell balance in mice colonized with inflammatory bowel disease microbiotas. Proc. Natl Acad. Sci. USA 117, 21536–21545 (2020).
Zarrinpar, A. et al. Antibiotic-induced microbiome depletion alters metabolic homeostasis by affecting gut signaling and colonic metabolism. Nat. Commun. 9, 2872 (2018).
Vandeputte, D. et al. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 65, 57–62 (2016).
Zhai, B. et al. High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis. Nat. Med. 26, 59–64 (2020).
Stoma, I. et al. Compositional flux within the intestinal microbiota and risk for bloodstream infection with gram-negative bacteria. Clin. Infect. Dis. 73, e4627–e4635 (2021).
Taur, Y. et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin. Infect. Dis. 55, 905–914 (2012).
Stein-Thoeringer, C. et al. Lactose drives Enterococcus expansion to promote graft-versus-host disease. Science 366, 1143–1149 (2019).
Maghini, D. G. et al. Quantifying bias introduced by sample collection in relative and absolute microbiome measurements. Nat. Biotechnol. 42, 328–338 (2024).
Rojas-Tapias, D. F. et al. Inflammation-associated nitrate facilitates ectopic colonization of oral bacterium Veillonella parvula in the intestine. Nat. Microbiol. 7, 1673–1685 (2022).
Sarrabayrouse, G. et al. Fungal and bacterial loads: noninvasive inflammatory bowel disease biomarkers for the clinical setting. mSystems https://doi.org/10.1128/msystems.01277-01220 (2021).
Schmidt, T. S. et al. Drivers and determinants of strain dynamics following fecal microbiota transplantation. Nat. Med. 28, 1902–1912 (2022).
Abusleme, L. et al. Oral microbiome characterization in murine models. Bio Protoc. 7, e2655 (2017).
Taur, Y. et al. Reconstitution of the gut microbiota of antibiotic-treated patients by autologous fecal microbiota transplant. Sci. Transl. Med. 10, eaap9489 (2018).
Djukovic, A. et al. Lactobacillus supports Clostridiales to restrict gut colonization by multidrug-resistant Enterobacteriaceae. Nat. Commun. 13, 5617 (2022).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Gonzalez, A. et al. Qiita: rapid, web-enabled microbiome meta-analysis. Nat. Methods 15, 796–798 (2018).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90 (2018).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Thomas, A. M. et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 25, 667–678 (2019).
Machado, D. et al. Polarization of microbial communities between competitive and cooperative metabolism. Nat. Ecol. Evol. 5, 195–203 (2021).
Peled, J. U. et al. Microbiota as predictor of mortality in allogeneic hematopoietic-cell transplantation. N. Engl. J. Med. 382, 822–834 (2020).
Nguyen, C. L. et al. High-resolution analyses of associations between medications, microbiome, and mortality in cancer patients. Cell 186, 2705–2718.e17 (2023).
Copelan, E. et al. A scheme for defining cause of death and its application in the T cell depletion trial. Biol. Blood Marrow Transplant. 13, 1469–1476 (2007).
Siranosian, B. A. et al. Rare transmission of commensal and pathogenic bacteria in the gut microbiome of hospitalized adults. Nat. Commun. 13, 586 (2022).
Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 11, 1144–1146 (2014).
Sieber, C. M. et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3, 836–843 (2018).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Brown, C. T., Olm, M. R., Thomas, B. C. & Banfield, J. F. Measurement of bacterial replication rates in microbial communities. Nat. Biotechnol. 34, 1256–1263 (2016).
Acknowledgements
This work was supported by National Institutes of Health (NIH) grant nos. U01 AI124275 (J.B.X.), R01 AI137269 (J.B.X.), R01 AI139632 (T.M.H.), R37 AI093808 (T.M.H.) and R21 AI156157 (T.M.H.). T.R. was funded by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) grant no. RO-5328/1-2. J.U.P. reports funding from NHLBI NIH Award K08HL143189, the MSKCC Cancer Center Core Grant NCI P30 CA008748.
Author information
Authors and Affiliations
Contributions
C. Liao, T.R., T.M.H. and J.B.X. conceptualized the project. A.D., V.M. and C. Lindberg performed the mouse experiment. C. Liao, A.D. and H.L. performed microbiome data processing. C. Liao, T.R. and T.F. conducted microbiome data analysis. C. Liao, T.R., A.D., H.L. and T.F. wrote the original draft. T.M.H., J.B.X., J.U.P., B.Z., L.D. and M.R.M.v.d.B. reviewed and edited the paper. J.B.X. and T.M.H. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
J.U.P. reports research funding, intellectual property fees and travel reimbursement from Seres Therapeutics, and consulting fees from DaVolterra, CSL Behring, Crestone, Inc. and Maat Pharma. He serves on an Advisory board of and holds equity in Postbiotics Plus Research. He has filed intellectual property applications related to the microbiome (reference numbers 62/843,849, 62/977,908 and 15/756,845). M.R.M.v.d.B. has received research support from Seres Therapeutics; he has consulted, received honorarium from or participated in advisory boards for Seres Therapeutics, WindMIL Therapeutics, Rheos, Frazier Healthcare Partners, Nektar Therapeutics, Notch Therapeutics, Forty Seven Inc., Priothera, Ceramedix, Lygenesis, Pluto Immunotherapeutics, Magenta Therapeutics, Merck & Co., Inc. and DKMS Medical Council (Board); and he has IP Licensing with Seres Therapeutics and Juno Therapeutics and stock options from Seres and Notch Therapeutics. T.M.H. has participated in a scientific advisory board for Boehringer-Ingelheim Inc. T.R. is currently an employee of BioNTech SE. The Memorial Sloan Kettering Cancer Center (MSKCC) has financial interests relative to Seres Therapeutics. The other authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Koji Atarashi, Christopher Staley and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Paired fecal and oral microbiome samples in the mouse experiments.
a, Bacterial ASV profiles displayed using stacked bar plots. Samples were collected at three time points: pre (before treatment), d3 (3 days after treatment initiation), and w1 (one week after treatment initiation). Missing samples in the second experiment were due to low sequence coverage (samples with less than 1,000 reads were excluded). b, Total bacterial load in fecal samples (circles). Bar heights represent the means, with error bars indicating the 95% confidence interval. P values were calculated using a one-sided Wilcoxon signed-rank test. DSS: Dextran Sulfate Sodium.
Extended Data Fig. 2 Quantitative agreement between our method and FEAST18 for estimating oral bacterial fraction in post-treatment fecal samples.
The FEAST-estimated values were averaged over 10 different runs. DSS: Dextran Sulfate Sodium. See Supplementary Note 1 for discussion on the discrepancy between the two approaches when oral bacterial fractions estimated by our approach fall below 0.0001.
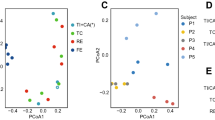
Extended Data Fig. 3 Principal coordinate analysis plot of the microbiome samples in the mouse experiment.
PCo1 and PCo2 represent principal component 1 and 2, respectively. a, Fecal and oral samples together. The unit of total bacterial load is 16S copies per gram of feces for fecal samples and 16S copies per swab for oral samples. b, Fecal samples only. Panel b shows that PCo1 captures the gut microbiota changes after antibiotic treatment, while PCo2 captures the gut microbiota changes after DSS treatment. DSS: Dextran Sulfate Sodium.
Extended Data Fig. 4 Validation of oral bacterial ASVs identified from healthy individuals in patients with inflammatory bowel disease.
This cohort consists of 16 patients with Crohn’s disease (CD), 42 patients with ulcerative colitis (UC), and 43 healthy controls (HC). Notably, none of the participants had taken antibiotics within the last three months prior to the study entry. a, Impact of cutoff parameters on the estimated oral bacterial fraction in feces of these participants. We systematically varied cutoffs for the mean relative abundance (0.01%, 0.1%), prevalence (5%, 10%), and the definition of ASV presence (0.01%, 0.1%) in the computation of prevalence to establish the reference set of oral ASVs. For each reference set, oral ASVs in their feces were inferred through exact sequence matching. The default parameter combination used throughout the study is outlined in the red box. P values were calculated using a one-sided Mann-Whitney U test. b, Percentage of inferred oral ASVs in feces that are also found in paired saliva samples. c, Percentage of the total relative abundance of inferred oral ASVs in feces contributed by those found in paired saliva samples. Two HC samples with a zero oral bacterial fraction are not shown in all box plots. Box plots represent the median, 25th and 75th percentiles and whiskers represent the 95th and 5th percentiles.
Extended Data Fig. 5 Validation of piperacillin-tazobactam’s effect on enriching oral bacteria in feces using an independent pediatric allo-HCT cohort.
19 children (1–17 year old, 10.1 year old on average) were treated with either oral polymyxin-neomycin or oral piperacillin-tazobactam in the Leiden University Medical Center, Netherlands. Both medications were administered 10 days before transplantation until engraftment or 21 days after transplantation, whichever occurred later. Samples were grouped into four transplantation stages. FDR-corrected P values were calculated using a one-sided Mann-Whitney U test. Box plots represent the median, 25th and 75th percentiles and whiskers represent the 95th and 5th percentiles.
Extended Data Fig. 6 Inverse correlation between oral bacterial fraction and total bacterial load in fecal samples from MSKCC allo-HCT recipients.
Each dot in the plot represents a fecal sample. Samples with zero oral bacterial fraction, or with total bacterial load less than 1,000 16S copies per gram of feces, or collected 20 days before or 40 days after transplantation were excluded from the plot and the linear regression analysis. The number of remaining samples is 2,524. The red line represents the best linear fit, and the shading of the same color indicates its ± 95% confidence interval. The plot also displays the estimated regression slope and its standard error.
Extended Data Fig. 7 Impact of interindividual variability on the regression slope between oral bacterial fraction and total bacterial load.
We generated synthetic datasets using parameters estimated from the MSKCC allo-HCT cohort analyzed in Fig. 4 (a) and the Crohn’s disease cohort analyzed in Fig. 5 (b). In both panels, we systematically varied σF (Eq. 1) from 0 to 2 while co-varying σf (Eq. 2) to maintain a constant ratio of σf/σF. For each synthetic dataset, the regression slope was determined through linear regression between oral bacterial fraction and total bacterial load in the log-log space. The red and blue lines represent the mean slopes over 100 simulation runs, with the shaded regions of the same color indicating the standard deviations. Vertical dashed lines in the panel (a) and (b) mark the σF values estimated from the pre-antibiotic-prophylaxis samples in the allo-HCT cohort and from the healthy individuals in the Crohn’s disease cohort, respectively. At these σF values, the pure Marker hypothesis predicted that the regression slopes are −0.36 and −0.30. For detailed information on our simulation approach, please refer to the Methods section.
Extended Data Fig. 8 Cross-validation accuracy for classifying total bacterial load in fecal samples from MSKCC allo-HCT recipients.
A schematic illustration of our classification model is enclosed in the dashed box. Our model uses two threshold parameters, θo and θt, to convert the oral bacterial fraction and total bacterial load into binary categories, respectively. Given training data, we optimized the two cutoff parameters by minimizing the P value of Fisher’s exact test of independence, subject to the constraints \({\theta }_{o}\in [{10}^{-4},1]\) and \({\theta }_{t}\in [{10}^{3},{10}^{10}]\). The optimized θo was subsequently applied to predict high or low bacterial loads in the test set by comparing the oral bacterial fractions to θo. Simultaneously, we binarized the observed bacterial loads by comparing them to θt. Accuracy was assessed by comparing the predicted bacterial load categories to the observed bacterial load categories. In the boxplot, each dot corresponds to a single 5-fold cross-validation split and the random train-test split was repeated 50 times. Box plots represent the median, 25th and 75th percentiles and whiskers represent the 95th and 5th percentiles.
Extended Data Fig. 9 Distribution of oral bacterial fraction in fecal samples from MSKCC allo-HCT recipients.
a,b, Each dot represents a fecal sample, categorized by stool consistency (a) and fungal culturability (b). FDR-corrected P values were calculated using a two-sided Mann-Whitney U test. Box plots represent the median, 25th and 75th percentiles and whiskers represent the 95th and 5th percentiles.
Extended Data Fig. 10 Emergence of new oral ASVs in mouse feces after antibiotic treatment.
Each panel plots data from one mouse. Mouse Abx_2B was excluded due to the absence of its pre-treatment fecal sample. In each panel, individual oral ASVs are shown by lines. ASVs that increased in absolute abundance after treatment (that is, post-treatment average > pre-treatment) are shown in red, while those that decreased or remained unchanged (post-treatment average ≤ pre-treatment) are shown in blue. The black line in each panel represents the total absolute abundance, which is the sum of all red and blue lines. Except for one oral ASV in mouse Abx_1B (shown as a red dashed line), all ASVs displaying increased absolute abundance (solid red lines) were not present in the pre-treatment fecal samples; they represent new ASVs emerging from the oral cavity. Samples were collected at three time points: pre (before treatment), d3 (3 days after treatment initiation), and w1 (one week after treatment initiation).
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2, Tables 2, 5 and 8, captions for Extended Images, Notes and References.
Supplementary Tables
Excel file with 9 worksheets (Supplementary Tables 1a–c, 3, 4a,b, 6, 7 and 9).
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liao, C., Rolling, T., Djukovic, A. et al. Oral bacteria relative abundance in faeces increases due to gut microbiota depletion and is linked with patient outcomes. Nat Microbiol (2024). https://doi.org/10.1038/s41564-024-01680-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41564-024-01680-3
