
Abstract
Herein, we develop a straightforward, metal-free, and acid-/base-free electrochemical C4-selective C − H deuteration of pyridine derivatives with economic and convenient D2O at room temperature. This strategy features an efficient and environmentally friendly approach with high chemo- and regioselectivity, affording a wide range of D-compounds, such as pyridines, quinolones, N-ligands and biorelevant compounds. Notably, the mechanistic experiments and cyclic voltammetry (CV) studies demonstrate that N-butyl-2-phenylpyridinium iodide is a crucial intermediate during the electrochemical transformation, which provides a general and efficient way for deuteration of pyridine derivatives.
Similar content being viewed by others


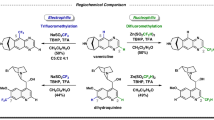
Introduction
Deuterium-labelled molecules are a critical kind of organic compounds, which have been widely applied into various research areas, such as elucidating the reaction mechanisms1,2, isotopic tracer techniques3,4,5, and pharmaceutical chemistry6,7,8,9,10. For example, N-heteroarenes, which are frequently employed as bioactive molecules and drugs for exploring the pharmacokinetic and pharmacodynamic (PK/PD) properties11,12. Therefore, it is unsurprising that the synthesis and application of deuterium-labeled (N-hetero)arenes has attracted significant attentions13,14,15,16,17,18, especially for the D-labeled pyridine derivatives19,20,21. Among the various protocols that have been developed, the direct, simple and efficient H/D exchange strategy22,23,24 stands prior to dehalogenative deuteration of halides and pseudohalides25,26,27, owing to the readily available starting materials, their cost, as well as high atom economy, however, the challenge would be the selectivity control of the transformations. Despite the established C−H deuteration of pyridine derivatives in the presence of Brønsted/Lewis acid28,29, base30,31, or transition-metal32,33 (Fig. 1A), the pursuance of methodologies with higher selectivity and D-incorporation under milder conditions with easy operation procedures is still on the way. Notably, McNally and co-workers developed a C4-selective C−H deuteration of pyridine derivatives through two-step process, which undergoing the heterocyclic phosphonium salts intermediator, and then forming the final deterateted pyridine derivatives with assistance of base, achieving high selective deuteration of pyridine derivatives (Fig. 1B)34. Furthermore, the development of approaches for site-selective C−H deuteration of pyridine derivatives in a straightforward, sustainable, and efficient way is still highly desirable and challenging.

A Previous methods for C−H deuteration of pyridine derivatives. B C4-selective two-step deuteration of pyridine derivatives via phosphonium salts. C This work: electrochemical C−H deuteration of pyridine derivatives.
In recent years, the emergence of electrochemistry has brought opportunities and development in organic synthesis35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52. Electrosynthesis is becoming increasingly popular and considered as one of sustainable and desirable methodology that could substitute some traditional synthetic methods. We envisage that direct and selective C−H deuteration of pyridine derivatives can be achieved through the usage of electrochemistry. However, compared with well-developed electrooxidative C−H functionalization53,54,55,56,57,58,59,60,61,62,63,64,65, electroreductively driven C−H functionalization of arenes have thus not been well elucidated66,67,68,69. Especially, selective C−H deuteration of pyridine derivatives under electroreductive conditions has not achieved yet.
In sharp contrast, with our continuous interests in sustainable electroreductively driven C−H functionalization66, and electrochemical deuteration70,71, herein, we would like to report our effort in developing a general, direct and efficient electrochemical C4-selective C−H deuteration of pyridine derivatives through reductive activation with economical D2O, via a crucial N-butyl-2-phenylpyridinium iodide intermediate (Fig. 1C). This protocol offered a wide range of D-pyridine derivatives with high chemo- and regioselectivity in excellent yields. The salient features of this transformation including: (a) electroreductively driven C−H deuteration; (b) good to excellent D-incorporation; (c) metal-, acid-, or base-free process; (d) high chemo- and regioselectivity; (e) late-stage modification of N-ligands and biorelevant compounds.
Results
Optimization of reaction conditions
Our investigations began by evaluating the C−H deuteration of pyridine substrates with deuterium oxide (D2O). After some preliminary experiments, 2-phenylpyridine (S1) was selected as the model starting substrate for optimizing the reaction conditions. Encouragingly, only a single product 1 was observed, which proved that the reaction has high regioselectivity. After a careful selection of the system parameters, we obtained the optimal conditions for deuteration of S1 with D2O. Combining the reactants with electrolyte nBu4NI in an undivided cell with DMF under constant current (20 mA) at room temperature for 10 hours, resulted in 99% yield and >99% deuterium incorporation (abbreviated as D-inc hereafter) (Table 1, entry 1). Initially, a variety of electrolytes were investigated. nBu4NBF4 and Et4NI worked well and resulted in 99% and 90% D-inc of product 1 respectively (Table 1, entries 2 and 3), but no product was detected when LiClO4 or NaI was employed (Table 1, entries 4 and 5). Then, an attempted to use NaOAc as a base, led to a decrease in deuteration (Table 1, entry 6). We also examined various solvents, such as DMA and MeCN, which afforded 82% and <5% D-inc of product 1 respectively (Table 1, entries 7 and 8). Next, we investigated various electrodes, including CF(+)|Pb(−), GF(+)|GF(−), and GF(+)|Pt(−) (Table 1, entries 9−11). However, the D-inc were <80%. Furthermore, we probed the effect of the current on this reaction. With a lower current, deuteration of 1 decreased significantly (Table 1, entries 12−13). The system also worked smoothly at higher temperatures (Table 1, entry 14, 50 °C). In addition, we found that this system was insensitive to the atmosphere and still gave excellent yield and D-inc under argon atmosphere (Table 1, entry 15). Finally, some control experiments proved that the presence of electricity and electrolyte were essential for this transformation (Table 1, entries 16 and 17).
Substrate scope
With optimal conditions in hand, we subsequently investigated the substrate scope and generality of this efficient electrochemical C−H deuteration transformation. As shown in Fig. 2A, a range of aryl-/alkyl-pyridines with diverse substituents were evaluated. The steric effect of the substituent exerted little impact on this transformation, for example, 2-phenylpyridines bearing o-, m-, or p-methyl substituent furnished the desired products in good to excellent yields and D-inc (2−4), as well as that bearing two methyl (5). Other substituents, such as -OMe (6), -tBu (7), -Ph (8), -OiPr (9), and -OPh (10) were tolerated smoothly. The substrates that bearing fluorine (11), trifluoromethyl (12) and carboxy (13) groups were compatible and worked successfully, providing the desired products with high efficiency. Moreover, the deuterated products from phenylpyridine with heteroaryl-based (S, O, N) groups were also obtained in excellent yields and D-inc (14−17). Moreover, alternate and multiple substituted substrates were all tolerated and reacted smoothly in the system, providing satisfactory results (18−22, up to 99% yields, 86%−> 99% D-inc). In addition to aryl pyridines, we also investigated diverse alkyl pyridine derivatives. A wide range of substrates with various electron-donating and electron-withdrawing groups could all react well with D2O and produce the corresponding deuterium products in 75% to >99% D-inc (23–28).

A Pyridine derivatives. B Quinolones. Reaction conditions: aElectrochemical C−H deuteration of pyridines and quinolones in an undivided cell, GF as anode and Pb as cathode, constant current (20 mA), pyridine derivatives (0.3 mmol), D2O (15.0 mmol), nBu4NI (1.0 equiv.), DMF (4.0 mL), room temperature, 10 h, isolated yield. Deuterium incorporation percentages were determined by 1H NMR spectroscopy. bThe reaction was conducted under 25 mA constant current. c30 mA. d40 mA. e16 h.
Quinoline is a crucial class of heterocycles that are widely found in natural products, pharmaceuticals, dyes, and materials72,73,74,75. Therefore, we tried to probe the scope of quinolines with diverse functional groups to further exhibit the applicability of this protocol (Fig. 2B). We found that quinolines bearing electron-neutral (29), -donating (30−39), and -withdrawing (40) functional groups performed well in this transformation, affording the corresponding products in good yields (94%–99%) with excellent D-inc (84%– > 99%) and selectivity. In addition, 7,8-Benzoquinoline (41) and acridine (42, vaccines against infection and allergy) were also successfully worked in this protocol, resulting in satisfying results.
More importantly, we turned our attention to N-ligands (Fig. 3A), various of them were also accommodated, leading to the desired molecules in both good yields and D-inc (43−50), including bipyridine (43−47, 45, Abametapir, a pediculicide for head lice infestation), benzimidazole (48) and phenanthroline (49−50), which indicated great compatibility and practicability of the protocol. It provided a new method for late-stage functional modification of N-ligands.

A N-ligands. B Biorelevant compounds. Reaction conditions: aElectrochemical C−H deuteration of pyridines and quinolones in an undivided cell, GF as anode and Pb as cathode, constant current (20 mA), pyridine derivatives (0.3 mmol), D2O (15.0 mmol), nBu4NI (1.0 equiv.), DMF (4.0 mL), room temperature, 10 h, isolated yield. Deuterium incorporation percentages were determined by 1H NMR spectroscopy. bThe reaction was conducted under 25 mA constant current. c30 mA. d40 mA. e16 h.
We next explored the late-stage deuteration of biorelevant compounds and some pharmaceutical molecules with this electroreductive method (Fig. 3B). Gratifyingly, pyridine compounds derived from Citronellol (51), Phytol (52), Borneol (53), l-menthol (54), Carveol (55), d-galactopyranose (56), Picaridin (57), (S)-N-Boc-2-hydroxymethylmorpholine (58), Ibuprofen (59) and some pharmaceutical molecules, including Loratadine (60, Claritin, an anti-allergic drug), Abiraterone acetate (61, Zytiga, a prostate cancer drug) and Bisacodyl (62, Dulcolax, a laxative), all showed great compatibility and reactivity with this electro-reductive system, delivering the desired deuterated products in both excellent yields and D-inc%. These results indicated that this electrochemical protocol has great potential and prospect in application and modification of biorelevant compounds.
Mechanistic studies
In order to further verify the rationality of this transformation, we performed a series of experiments to investigate the mechanism (Fig. 4). Firstly, we explored the effect of electrolytes (Fig. 4A). As expected, no product 1 was observed when NaCl, LiBF4 or KPF6 was employed as the electrolyte (Fig. 4A, entries 1–3). However, when we used other electrolytes containing nBu4N+ ion, excellent D-inc of 1 was obtained (Fig. 4A, entries 4–9). Meanwhile, various anions from salts led to different yields (yield of 1 or recovered S1). These results illustrated that nBu4N+ ion was crucial for this electrochemical reaction. Moreover, under the optimized conditions, N-butyl-2-phenylpyridinium iodide (S1-a, detected by HRMS, 212.1434), nBuI and nBu3N were afforded in the absence of D2O, which further demonstrated the important role of nBu4N+ (Fig. 4B). Furthermore, we speculated that S1-a might be a key intermediate in this process. Hence, nBu3N (1.0 equiv.) instead of nBu4NI, then a catalytic amount (5 mol%) of S1-a~S1-d were added to 2-phenylpyridine (S1) under standard conditions, furnishing the product 1 with >99% D (S1-a, nBu+), 96% D (S1-b, Pr+), 90% D (S1-c, Et+) and 87% D (S1-d, Me+) respectively (Fig. 4C, a). On the other hand, the corresponding electrolytes were employed for the model reaction with standard conditions and gave compound 1 with >99% D (nBu4NI), 95% D (Pr4NI), 90% D (Et4NI), and 87% D (Me4NI) severally (Fig. 4C, b). The results were almost identical to the previous ones. Next, we conducted several cyclic voltammetry (CV) studies (for details, please see the Supplementary Information on page 32). CV experiments on N-butyl-2-phenylpyridinium iodide (S1-a) gave a reductive peak at Ep/2 =−1.71 V (−0.401 mA) vs. Ag/Ag+ under Ar atmosphere (Fig. 4D, a, green line). An obvious reversible oxidative peak at −2.65 V (0.246 mA) vs. Ag/Ag+ and a reversible reductive peak of S1 at −3.14 V (−1.038 mA) vs. Ag/Ag+ were observed under Ar atmosphere (Fig. 4D, a, red line). After mixing the two ingredients, the reductive peak of S1-a moved to −1.70 V (−0.453 mA) vs. Ag/Ag+ (Fig. 4D, a, purple line). The oxidative and reductive peaks of S1 changed to −2.68 V (0.142 mA) vs. Ag/Ag+ and −3.19 V (−1.474 mA) vs. Ag/Ag+ respectively (Fig. 4D, a, purple line). When D2O was added, the reductive peak of S1-a reduced to −1.68 V (−0.254 mA) vs. Ag/Ag+ (Fig. 4D, b, blue line). The reductive peak of the mixture changed from −3.19 V (−1.474 mA) to −3.27 V (−1.766 mA) vs. Ag/Ag+ under Ar atmosphere (Fig. 4D, b, blue line). However, the oxidative peak of the mixture was fully disappeared (Fig. 4D, b, blue line). All the results showed that a catalytic current was generated because of S1-a, which promoted the success of the protocol. Additionally, control experiments and CV studies also proved the bifunctional participation of nBu4NI including the improvement of conductivity and the synthesis of S1-a in this protocol.

A Effect of the electrolyte. B Observation of intermediate S1-a. C Contrast experiments. D Cyclic voltammetry studies, using glass carbon as work electrode, Pt plate and Ag/Ag+ as counter and reference electrodes. Scan rate: 100 mV s−1. Solvent: DMF/nBu4NBF4 (0.1 M) or MeCN/KPF6 (0.1 M), S1 (0.01 M), D2O (0.5 M), S1-a (0.001 M). Experiments were conducted under Ar unless otherwise noted. a CVs of S1-a, S1 and the mixture of them. b CVs of S1, S1 with S1-a and both of them with D2O. E 100 mmol scale reaction. F Flow-synthesis reaction. G D/H exchange experiments. H Competition experiments.
To show the robustness and utility of this reaction, we attempted to carry out a 10 mmol gram-scale experiment and a 100 mmol scale experiment to synthesize the product 1 (Fig. 4E). Surprisingly, the reaction maintained good selectivity with excellent yield and D-inc% (14.2 g, 91%, >99%D), which illustrated the high superiority and efficiency of this electroreductive protocol (For details, please see the Supplementary Information on pages 14−15). Meanwhile, we realized an elctrochemical continuous-flow reaction using a flow rate of 0.6 mL/min and a residence time for 3 h. The product 1 was obtained in excellent yield and D-inc% (Fig. 4F), which further demonstrated the potential application of this transformation. Then, we used several deuterated products to perform D/H exchange experiments with H2O (Fig. 4G), as shown, all D-molecules provided the corresponding initial materials in high yields (Fig. 4G, S1, S6, S8, S16, S19 and S39, 95% H−> 99% H). It indicated that this electrochemical C−H deuteration transformation was reversible. Next, several competition experiments were performed (Fig. 4H). When this system was carried out in a mixture of D2O/H2O (1/1, 7.5 mmol/7.5 mmol), only 16 % D of product 1 was produced (Fig. 4H, a), when the S1 was replaced by 1 under the same conditions, 24 % D-inc% of 1 was afforded (Fig. 4H, b), which demonstrated that the ability of pyridine anions to capture H+ is better than D+. In addition, more experiments were conducted to explore the H/D exchange and D/H exchange rate (For details, please see the Supplementary Information on pages 18−20). In addition, a mixture of two substrates bearing diverse substituents performed different results under the same conditions (Fig. 4H, c, d). For example, it was obvious that the D-inc % of products that bearing -F was superior to that bearing -OMe.
Cyclic voltammetry studies
To gain further exploration into the reaction mechanism, in-depth studies were carried out through detailed cyclic voltammetry studies. An obvious reversible oxidation peak at −2.65 V (0.238 mA) vs. Ag/Ag+ and a reversible reduction peak of S1 at −3.14 V (−1.025 mA) vs. Ag/Ag+ were observed under Ar atmosphere (Fig. 5, red line). In the presence of D2O, the reversible oxidation peak at −2.65 V (0.238 mA) vs. Ag/Ag+ disappeared and the reduction peak shifted from −3.14 V (−1.025 mA) to −3.18 V (−1.204 mA) vs. Ag/Ag+ (Fig. 5, blue line). In the mixture of S1 and nBu4NI, the reductive current of S1 increased slightly, which might be attributed to the slight variation in conductivity (Fig. 5, green line). Then, D2O was added to the mixed solution and the oxidation peak of S1 disappeared again (Fig. 5, dark blue line), which was consistent with the previous results. However, since no desired product 1 was detected with standard conditions in the absence of nBu4NI, it illustrated that nBu4NI played a crucial role in promoting the reactivity (Table 1, entry 17). Notably, we conducted CV experiments on S1 with various scan rates (Fig. 5c), and the linear fit analysis also explained that a diffusion-control process might be involved in the conversion (Fig. 5d).

Using glass carbon as work electrode, Pt plate and Ag/Ag+ as counter and reference electrodes. Scan rate: 100 mV s−1. Solvent: DMF/nBu4NBF4 (0.1 M) or MeCN/KPF6 (0.1 M), S1 (0.01 M), D2O (0.5 M), nBu4NI (0.01 M). Experiments were conducted under Ar unless otherwise noted. a CVs of S1 and S1 with D2O. b CVs of S1, S1 with nBu4NI and the reaction system. c CVs of S1 performed at variable scan rates ranging from 10 mV s−1 to 100 mV s−1. d Linear fit analysis of vscan1/2 and ip.
Based on the mechanistic experiments and CV studies, we proposed a possible mechanism (Fig. 6). Firstly, nBu4NI (I) splits into nBu3N (II) and nBuI (III). Then nBu3N (II) was oxidized to radical cation IV on the anode to offer electrons. Next, nBuI (III) forms a complex with S1 to afford intermediate V (S1-a). Subsequently, V (S1-a) undergoes a single electron transfer on the cathode, to form the radical intermediate VI. Another single electron reduction generates the anion intermediate VII. The unique and high regioselectivity might be determined by these procedures. Subsequently, the anion intermediate VII reacts with D2O and produces the deuteration intermediate VIII. Finally, intermediate VIII was oxidized on the anode, affording the target product 1.

Anode: oxidative reaction. Cathode: reductive reaction. S1 (2-phenylpyridine).
Discussion
In conclusion, we reported the direct and efficient C4-selective deuteration of pyridine derivatives via the mode of electroreductively driven C−H functionalization with D2O at room temperature, without any metal, acid, and base. This transformation proceeded smoothly, as demonstrated with a wide range of substrates, forming the desired products with high regioselectivity and good to excellent D-incorporation. The utility of this protocol was also shown in the synthesis of deuterated N-ligands and late-stage modification of biorelevant compounds. Moreover, the mechanistic experiments and CV studies showed that N-butyl-2-phenylpyridinium iodide promotes the success of the electroreductive C−H deuteration procedure. Further electrochemical transformation and applications of pyridine salts and derivatives are ongoing in our laboratory.
Methods
General procedure of electroreductive C−H deuteration of pyridine derivatives
The electrocatalysis was carried out in an undivided cell with graphite felt (GF, 10 mm × 15 mm × 5 mm) as anode and Pb (10 mm × 15 mm × 0.3 mm) as cathode. To an oven-dried undivided electrochemical cell (15 mL) equipped with a magnetic bar was added organic N-heteroarenes (0.3 mmol, 1.0 equiv.), nBu4NI (0.3 mmol, 110.8 mg, 1.0 equiv.) and D2O (15 mmol, 300 mg, 50.0 equiv.), then anhydrous DMF (4.0 mL) was added via a syringe. The electrocatalysis system was performed at 20–40 mA of constant current for 10 h at room temperature. After that, the reaction mixture was extracted with EtOAc (30 mL × 3) and the combined organic phase was dried by anhydrous MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography to furnish the deuterated products.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files. Extra data are available from the author upon request. Source data are provided with this paper.
References
Simmons, E. M. & Hartwig, J. F. On the interpretation of deuterium kinetic isotope effects in C−H bond functionalizations by transition-metal complexes. Angew. Chem. Int. Ed. 51, 3066–3072 (2012).
Giagou, T. M. & Meyer, P. Kinetic isotope effects in asymmetric reactions. Chem. Eur. J. 16, 10616–10628 (2010).
Kao, C. & Giese, R. W. Measurement of N7-(2‘-hydroxyethyl)guanine in human DNA by gas chromatography electron capture mass spectrometry. Chem. Res. Toxicol. 18, 70–75 (2005).
Atzrodt, J. & Derdau, V. Pd- and Pt-catalyzed H/D exchange methods and their application for internal MS standard preparation from a Sanofi-Aventis perspective. J. Label. Compd Radiopharm. 53, 674–685 (2010).
Allais, C., Grassot, J. M., Rodriguez, J. & Constantieux, T. Metal-free multicomponent syntheses of pyridines. Chem. Rev. 114, 10829–10868 (2014).
Katsnelson, A. Heavy drugs draw heavy interest from pharma backers. Nat. Med. 19, 656 (2013).
Mullard, A. Deuterated drugs draw heavier backing. Nat. Rev. Drug Discov. 15, 219–221 (2016).
Mullard, A. FDA approves first deuterated drug. Nat. Rev. Drug Discov. 16, 305 (2017).
Pirali, T., Serafini, M., Cargnin, S. & Genazzani, A. A. Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276–5297 (2019).
Schmidt, C. First deuterated drug approved. Nat. Biotechnol. 35, 493–494 (2017).
Sanderson, K. Big interest in heavy drugs. Nature 458, 269 (2009).
Nag, S. et al. Development of a novel fuorine-18 labeled deuterated fluororasagiline ([18F]Fluororasagiline-D2) radioligand for PET studies of monoamino oxidase B (MAO-B). Bioorg. Med. Chem. 21, 6634–6641 (2013).
Kopf, S. et al. Recent Developments for the deuterium and tritium labeling of organic molecules. Chem. Rev. 122, 6634–6718 (2022).
Derdau, V., Atzrodt, J., Zimmermann, J., Kroll, C. & Brückner, F. Hydrogen–deuterium exchange reactions of aromatic compounds and heterocycles by NaBD4-activated rhodium, platinum and palladium catalysts. Chem. Eur. J. 15, 10397–10404 (2009).
Pieters, G. et al. Regioselective and stereospecific deuteration of bioactive aza compounds by the use of ruthenium nanoparticles. Angew. Chem. Int. Ed. 53, 230–234 (2014).
Tlahuext, A. A. & Hartwig, J. F. Site-selective silver-catalyzed C–H bond deuteration of five-membered aromatic heterocycles and pharmaceuticals. ACS Catal. 11, 1119–1127 (2021).
Li, W. et al. Scalable and selective deuteration of (hetero)Arenes. Nat. Chem. 14, 334–341 (2022).
Farizyan, M., Mondal, A., Mal, S., Deufel, F. & van Gemmeren, M. Palladium-catalyzed nondirected late-stage C–H deuteration of arenes. J. Am. Chem. Soc. 143, 16370–16376 (2021).
Zarate, C., Yang, H., Bezdek, M. J., Hesk, D. & Chirik, P. J. Ni(I)–X complexes bearing a bulky α-diimine ligand: synthesis, structure, and superior catalytic performance in the hydrogen isotope exchange in pharmaceuticals. J. Am. Chem. Soc. 141, 5034–5044 (2019).
Daniel-Bertrand, M. et al. Multiple site hydrogen isotope labelling of pharmaceuticals. Angew. Chem. Int. Ed. 59, 21114–21120 (2020).
Mai, V. H., Gadzhiev, O. B., Ignatov, S. K. & Nikonov, G. I. H/D exchange in N-heterocycles catalysed by an NHC-supported ruthenium complex. Catal. Sci. Technol. 9, 3398–3407 (2019).
Junk, T. & Catallo, W. J. Hydrogen isotope exchange reactions involving C−H (D, T) bonds. Chem. Soc. Rev. 26, 401–406 (1997).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. Deuterium- and tritium-labelled compounds: applications in the life sciences. Angew. Chem. Int. Ed. 57, 1758–1784 (2018).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. C–H functionalisation for hydrogen isotope exchange. Angew. Chem. Int. Ed. 57, 3022–3047 (2018).
Liu, C. et al. Controllable deuteration of halogenated compounds by photocatalytic D2O splitting. Nat. Commun. 9, 80 (2018).
Liu, C., Han, S., Li, M., Chong, X. & Zhang, B. Electrocatalytic deuteration of halides with D2O as the deuterium source over a copper nanowire arrays cathode. Angew. Chem. Int. Ed. 59, 18527–18531 (2020).
Li, Y. et al. Organophotocatalytic selective deuterodehalogenation of aryl or alkyl chlorides. Nat. Commun. 12, 2894 (2021).
Garnett, J. L., Long, M. A., Vining, R. & Mole, T. New simple method for rapid, selective aromatic deuteration using organoaluminum dihalide catalysts. J. Am. Chem. Soc. 94, 5913–5914 (1972).
Martins, A., Lautens, M. & Simple, A. A simple, cost-effective method for the regioselective deuteration of anilines. Org. Lett. 10, 4351–4353 (2008).
Zoltewicz, J. A., Grahe, G. & Smith, C. L. Unusual influence of nitrogen on rates of anion formation. Hydrogen-deuterium exchange of pyridine and the diazines. J. Am. Chem. Soc. 91, 5501–5505 (1969).
Kebede, N. & Pavlik, J. W. Hydrogen-deuterium exchange in dimethylpyridines. J. Heterocycl. Chem. 34, 685–686 (1997).
Alexakis, E., Jones, J. R. & Lockley, W. J. S. One-step exchange-labelling of pyridines and other N-heteroaromatics using deuterium gas: catalysis by heterogeneous rhodium and ruthenium catalysts. Tetrahedron Lett. 47, 5025–5028 (2006).
Yang, H. et al. Site-selective Nickel-catalyzed hydrogen isotope exchange in N-Heterocycles and its application to the tritiation of pharmaceuticals. ACS Catal. 8, 10210–10218 (2018).
Koniarczyk, J. L. et al. A general strategy for site-selective incorporation of deuterium and tritium into pyridines, diazines, and pharmaceuticals. J. Am. Chem. Soc. 140, 1990−1993 (2018).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: On the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Wiebe, A. et al. Electrifying organic synthesis. Angew. Chem. Int. Ed. 57, 5594–5619 (2018).
Gandeepan, P., Finger, L. H., Meyer, T. H. & Ackermann, L. 3d metallaelectrocatalysis for resource economical syntheses. Chem. Soc. Rev. 49, 4254–4272 (2020).
Novaes, L. et al. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 50, 7941–8002 (2021).
Jutand, A. Contribution of electrochemistry to organometallic catalysis. Chem. Rev. 108, 2300–2347 (2008).
Yoshida, J., Kataoka, K., Horcajada, R. & Nagaki, A. Modern strategies in electroorganic synthesis. Chem. Rev. 108, 2265–2299 (2008).
Francke, R. & Little, R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 43, 2492–2521 (2014).
Yuan, Y. & Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution reactions. Acc. Chem. Res. 52, 3309–3324 (2019).
Xiong, P. & Xu, H. Chemistry with electrochemically generated N-centered radicals. Acc. Chem. Res. 52, 3339–3350 (2019).
Kingston, C. et al. A survival guide for the “Electro-Curious”. Acc. Chem. Res. 53, 72–83 (2020).
Wang, F. & Stahl, S. Electrochemical oxidation of organic molecules at lower overpotential: accessing broader functional group compatibility with electron-proton transfer mediators. Acc. Chem. Res. 53, 561–574 (2020).
Röckl, J. L., Pollok, D., Franke, R. & Waldvogel, S. R. A decade of electrochemical dehydrogenative C, C-coupling of aryls. Acc. Chem. Res. 53, 45–61 (2020).
Siu, J. C., Fu, N. & Lin, S. Catalyzing electrosynthesis: a homogeneous electrocatalytic approach to reaction discovery. Acc. Chem. Res. 53, 547–560 (2020).
Shi, S., Liang, Y. & Jiao, N. Electrochemical oxidation induced selective C–C bond cleavage. Chem. Rev. 121, 485–505 (2021).
Liu, Y., Li, P., Wang, Y. & Qiu, Y. Electroreductive cross-electrophile coupling (eXEC) reactions. Angew. Chem. Int. Ed. 62, e202306679 (2023).
Liu, C., Wu, Y., Zhao, B. & Zhang, B. Designed nanomaterials for electrocatalytic organic hydrogenation using water as the hydrogen source. Acc. Chem. Res. 56, 1872–1883 (2023).
Wang, Y.-W., Wang, Q. & Qiu, Y.-A. Electroreduction of unactivated alkenes using water as hydrogen source. Nat. Commun. 15, 2780 (2024).
Li, P.-F., Zhu, Z.-L. & Qiu, Y.-A. Nickel-electrocatalysed C(sp3)–C(sp3) cross-coupling of unactivated alkyl halides. Nat. Catal. https://doi.org/10.1038/s41929-024-01118-3 (2024).
Horn, E. J. et al. Scalable and sustainable electrochemical allylic C−H oxidation. Nature 533, 77–81 (2016).
Sauermann, N., Meyer, T. H., Tian, C. & Ackermann, L. Electrochemical cobalt-catalyzed C−H oxygenation at room temperature. J. Am. Chem. Soc. 139, 18452–18455 (2017).
Xiong, P. et al. Electrochemically enabled carbohydroxylation of alkenes with H2O and organotrifluoroborates. J. Am. Chem. Soc. 140, 16387–16391 (2018).
Liang, Y. et al. Electrochemically induced nickel catalysis for oxygenation reactions with water. Nat. Catal. 4, 116–123 (2021).
Shen, T. & Lambert, T. H. Electrophotocatalytic diamination of vicinal C−H bonds. Science 371, 620–626 (2021).
Chung, D. S., Park, S. H., Lee, S. G. & Kim, H. Electrochemically driven stereoselective approach to syn-1,2-diol derivatives from vinylarenes and DMF. Chem. Sci. 12, 5892–5897 (2021).
Chung, D. S., Park, S. H., Lee, S. G. & Kim, H. Enantioselective electrochemical cobalt-catalyzed aryl C–H activation reactions. Science 379, 1036–1042 (2023).
Shen, T., Li, Y. L., Ye, K. Y. & Lambert, T. H. Electrophotocatalytic oxygenation of multiple adjacent C–H bonds. Nature 614, 275–280 (2023).
Sauermann, N., Meyer, T. H., Qiu, Y. & Ackermann, L. Electrocatalytic C−H activation. ACS Catal. 8, 7086–7103 (2018).
Jiao, K.-J., Xing, Y.-K., Yang, Q.-L., Qiu, H. & Mei, T.-S. Site-selective C−H functionalization via synergistic use of electrochemistry and transition metal catalysis. Acc. Chem. Res. 53, 300–310 (2020).
Waldvogel, S. R., Lips, S., Selt, M., Riehl, B. & Kampf, C. J. Electrochemical arylation reaction. Chem. Rev. 118, 6706–6765 (2018).
Wang, H., Gao, X., Lv, Z., Abdelilah, T. & Lei, A. Recent advances in oxidative R1−H/R2−H cross-coupling with hydrogen evolution via photo-/electrochemistry. Chem. Rev. 119, 6769–6787 (2019).
Liu, M., Feng, T. & Qiu, Y. Metal-free electrochemical dihydroxylation of unactivated alkenes. Nat. Commun. 14, 6467 (2023).
Zhao, Z.-W. et al. Site-selective electrochemical C−H carboxylation of arenes with CO2. Angew. Chem. Int. Ed. 62, e202214710 (2023).
Sun, G.-Q. et al. Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations. Nature 615, 67–72 (2023).
Wang, M. et al. Room temperature construction of vicinal amino alcohols via electroreductive cross-coupling of N-heteroarenes and carbonyls. J. Am. Chem. Soc. 145, 10967–10973 (2023).
Wang, P. et al. Electrochemical arylation of electron-deficient arenes through reductive activation. Angew. Chem. Int. Ed. 58, 15747–15751 (2019).
Li, P. et al. Facile and general electrochemical deuteration of unactivated alkyl halides. Nat. Commun. 13, 3774 (2022).
Yang, K., Feng, T. & Qiu, Y. Organo-mediator enabled electrochemical deuteration of styrenes. Angew. Chem. Int. Ed. 62, e202312803 (2023).
Hughes, G. & Bryce, M. R. Electron-transporting materials for organic electroluminescent and electrophosphorescent devices. J. Mater. Chem. 15, 94–107 (2005).
Michael, J. P. Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 25, 166–187 (2008).
Mu, L., Shi, W., Chang, J. C. & Lee, S. T. Silicon nanowires-based fluorescence sensor for Cu(II). Nano Lett. 8, 104–109 (2008).
Solomon, V. R. & Lee, H. Quinoline as a privileged scaffold in cancer drug discovery. Curr. Med. Chem. 18, 1488–1508 (2011).
Acknowledgements
Financial support from the National Key R&D Program of China (2023YFA1507203), National Natural Science Foundation of China (Grant No. 22371149, 22188101), the Fundamental Research Funds for the Central Universities (No. 63223015), Frontiers Science Center for New Organic Matter, Nankai University (Grant No. 63181206), and Nankai University are gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
Y.Q. supervised the project, and provided guidance on the project. Y.Q. and Z.W.Z. conceived and designed the study and wrote the manuscript. Z.W.Z., R.Z., Y.L., Q.W. and Y.Q. performed the experiments, mechanistic studies, and revised the manuscript. Z.L.Z. conducted DFT calculations. All authors contributed to the analysis and interpretation of the data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications Hai-Chao Xu for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, Z., Zhang, R., Liu, Y. et al. Electrochemical C−H deuteration of pyridine derivatives with D2O. Nat Commun 15, 3832 (2024). https://doi.org/10.1038/s41467-024-48262-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-48262-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
