
Abstract
Transition metal-catalyzed enantioconvergent cross-coupling of an alkyl precursor presents a promising method for producing enantioenriched C(sp3) molecules. Because alkyl alcohol is a ubiquitous and abundant family of feedstock in nature, the direct reductive coupling of alkyl alcohol and aryl halide enables efficient access to valuable compounds. Although several strategies have been developed to overcome the high bond dissociation energy of the C − O bond, the asymmetric pattern remains unknown. In this report, we describe the realization of an enantioconvergent deoxygenative reductive cross-coupling of unactivated alkyl alcohol (β-hydroxy ketone) and aryl bromide in the presence of an NHC activating agent. The approach can accommodate substituents of various sizes and functional groups, and its synthetic potency is demonstrated through a gram scale reaction and derivatizations into other compound families. Finally, we apply our convergent method to the efficient asymmetric synthesis of four β-aryl ketones that are natural products or bioactive compounds.
Similar content being viewed by others
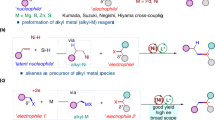

Introduction
The saturation degree and the presence of chiral centers are two factors that correlate to the successful transition of a compound from discovery, to clinical testing, and ultimately into a drug1. Transition metal-catalyzed enantioconvergent cross-coupling of an alkyl precursor presents a promising method for producing these molecules2,3,4,5,6,7. This approach has been shown to be highly effective in forging C(sp3)−C(sp2) bond, particularly when using nickel as the catalyst8,9,10,11,12,13,14,15,16,17,18. Traditional coupling utilizes alkyl halide and organometallic reagent to form a new C − C bond (Fig. 1a)19,20,21,22. The application of reductive cross-coupling, led by Weix23,24,25,26,27,28,29, Reisman30,31,32,33,34,35,36,37,38,39,40, and others41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64, has proven beneficial in circumventing the utilization of organometallic reagents that are vulnerable to air and moisture, and in shortening the synthesis with fewer steps. On the other hand, alkyl halide can be produced through the Appel reaction using alkyl alcohol. Alkyl alcohol is an abundant alkyl source in nature and would be a desirable choice for C(sp3) coupling. However, the direct cross-coupling of alkyl alcohols remains an underdeveloped field, especially when creating asymmetric patterns65,66,67,68,69.

a Comparison of different cross-coupling reactions. b A breakthrough from the MacMillan group. c Examples of natural products and bioactive molecules. d This study: Ni-catalyzed enantioconvergent deoxygenative reductive cross-coupling of unactivated alkyl alcohols and aryl bromides. Met metal complex, NHC N-aryl benzoxazolium salt, PC photocatalyst.
Due to the high bond dissociation energy of the C − O bond and the low leaving ability of the OH− group70, the direct reductive cross-coupling of alkyl alcohols with aryl halides is elusive. In this vein, many types of alcohol derivatives, such as alkyl acetates71,72, tosylates73,74,75, xanthate esters76, mesylates77, pivalates78,79, oxalates80,81,82, methyl ethers83, chloroformates84, and others85,86,87, have been extensively explored in reductive cross-coupling reactions. However, most of these methods are limited to activated alkyl alcohol derivatives, and the pre-activation requires additional steps. In 2018, Ukaji and co-workers offered an appealing solution by employing a Ti-mediated direct reductive cross-coupling, which only works with primary benzyl alcohols88,89,90,91,92,93,94,95,96,97,98. An alternative strategy that may be used in certain cases is the one-pot process involving the in situ activation of alkyl alcohols and their subsequent reductive cross-coupling. In this context, pioneering studies from Li99,100, Gong101, Weix102, Shu103,104,105,106, and others107,108,109,110,111,112,113 have demonstrated the power of this approach owing to the ubiquity of the two building blocks: free alcohols and aryl bromides. A breakthrough was disclosed by the MacMillan group, who realized an NHC (N-heterocyclic carbene) enabled deoxygenative arylation in 2021 (Fig. 1b)114,115,116,117,118,119. This robust method features mild conditions and simple operations, and targets a broad spectrum of primary, secondary, and tertiary alcohols.
Despite these significant advances, an enantioconvergent deoxygenative reductive cross-coupling of an alkyl alcohol, especially an unactivated alkyl alcohol, still needs to be addressed. Managing the stereoselective nature of free radical reactions remains a formidable task because of the high reactivity of radical species. β-Hydroxy ketones are a readily available building block that can be obtained by one-step aldol condensation and contain an unactivated alkyl alcohol group. Furthermore, the carbonyl group is an essential functional group in organic chemistry due to its versatility in forming a variety of structures and its prevalence in numerous biologically relevant compounds. We speculate that MacMillan’s robust NHC system would serve as an excellent foundation for achieving the enantioconvergent deoxygenative reductive cross-coupling of β-hydroxy ketones, providing ready access to a wide variety of β-aryl ketones that are a common subunit found in many natural products and bioactive molecules (Fig. 1c). Herein we describe the realization of this objective by using a chiral nickel/pyridyloxazoline catalyst (Fig. 1d).
Results
Reaction optimization
In an initial study, we examined the coupling of racemic 3-hydroxy-1-phenyl-1-heptanone with methyl 4-bromobenzoate (Table 1). We chose an N-aryl benzoxazolium salt (NHC) as the activator to convert the alkyl alcohol to an NHC-alcohol adduct in situ. It’s worth noting that this benzoxazolium salt can be easily prepared in the lab on a hundred-gram scale. After an extensive evaluation of all reaction parameters, we determined that NiBr2·DME and chiral pyridyloxazoline ligand L1 can accomplish the desired enantioconvergent deoxygenative reductive cross-coupling in good yield and ee (83% yield, 92% ee; entry 1).
In the absence of NiBr2·DME, photocatalyst, quinuclidine, light, or ligand L1, essentially no or only a small amount of product is observed (racemic; entries 2 and 3). The presence of 4-methylpyridine is crucial in the deoxygenative reductive coupling process. The absence of this additive results in a product with much lower efficiency and selectivity (entry 4). A variety of other chiral ligands are less effective than ligand L1 (entries 5–9). Moreover, the mixed solvent proves superior to a single solvent (entries 10 and 11). Employing any other base or additive results in a subpar outcome (entries 12 and 13). If the coupling is conducted with less catalyst, for a shorter time, or at an elevated temperature (r.t.), then a lower yield and/or ee are obtained (entries 14-16). The reaction proceeds relatively smoothly in the presence of a small amount of air (entry 17), whereas a reaction run with water leads to a diminished yield and ee (entry 18). Under these conditions, the corresponding ester, amide, and phosphonate are not suitable coupling partners (entry 19). Additionally, substrates like 2-butanol and others that have a functional group at the β-position (-Ph, -OBz, -NHCbz, -NHBz, -NPh2, etc.), were found to provide much lower yield and/or ee (for a broad exploration of other potential substrates, see Supplementary Fig. 6 and Supplementary Fig. 7), highlighting the critical role of the ketone moiety as a potential directing group in the cross-coupling reaction120. It is also noteworthy that the MacMillan group employed a similar pyridyloxazoline ligand to successfully couple alkyl alcohols and alkyl bromides in an achiral/racemic manner, which offers further support to the notion that the ketone moiety used in this study plays a significant role in enantiocontrol118.
Substrate scope
With the optimized reaction conditions in hand, we sought to examine the generality of substrate scope for both coupling partners. This straightforward method for the catalytic enantioconvergent synthesis of arylated products is compatible with an array of substituents at the β-position (R1; Fig. 2a) of the ketones, providing a range of products with good yields and high ee. For example, the alkyl substituent at the β-position can vary in size from methyl to neopentyl to isopropyl, and consistently good yields and ee are observed (products 1–7). A variety of functional groups can be present, including silyl ether, ether, ester, unactivated primary alkyl fluoride/chloride, terminal olefin, and Boc-protected amine (products 8−18). In reactions involving alcohols with a stereocenter, the catalyst determines the stereochemistry outcome, rather than the substrate (products 19 and 20). Notably, the presence of an aryl group at the β-position in place of an alkyl group results in a comparable outcome as well (products 21–23).

a Variations of substituents at the β-position of ketones. b Variations of substituents attached to the carbonyl of ketones. c Variations of aryl bromides. All couplings were conducted on a 0.50 mmol scale (unless otherwise noted), and all yields are of purified products. TBS tert-butyldimethylsilyl, Boc tert-butoxycarbonyl, Bpin pinacolato-boron.
With regard to the groups attached to the carbonyl of the ketones (R2; Fig. 2b), many aryls prove to be appropriate, including several heteroaryls such as furan, thiophene, and benzothiophene (products 24–36). Furthermore, not only aryl ketones but also alkyl ketones illustrate superior reactivity and selectivity, and the alkyl size can vary in size from methyl to tert-butyl (products 37–41).
We next evaluated the scope of aryl bromides (Ar; Fig. 2c). Under similar conditions, the chiral nickel catalyst couples 1.0 equivalent of racemic β-hydroxy ketone to provide the substitution product with good enantioselectivity and yield (for example, product 44, 70% yield, 93% ee). The observed values of the enantiomeric excess and yield provide evidence that the coupling reaction works as an enantioconvergent process. In this context, the catalyst efficiently converts both enantiomers of the racemic alkyl alcohol substrate into a specific stereoisomer of the desired product. This protocol can efficiently incorporate aryl bromides containing either electron-rich or electron-deficient substituents, complementing previously established Ni-catalyzed reductive cross-couplings that are typically limited to the electron-deficient aryl halides. Many functional groups, including ester, fluoride/chloride, trifluoromethyl, Bpin, nitrile, and ketone, are well tolerated in the current system (products 3, 42–55). Unfortunately, the reaction cannot be carried out with o-substituted aryl bromides (o-F and o-Me) due to the increased steric hindrance. Aryl rings can also be replaced by heteroaryls, including quinoline, benzofuran, and benzothiophene (products 56–58). In reactions involving aryl bromides with one or more stereocenters, the catalyst determines the stereochemistry outcome instead of the substrate (products 59−62). A reaction on a gram scale (1.22 g of product) yields coupling product 46 with similar yield and enantiomeric excess as observed in a reaction performed on a 0.50 mmol scale. The absolute configuration of products was unambiguously determined through X-ray diffraction analysis of compounds 21, 28, and 39.
Applications and mechanistic observations
To illustrate the synthetic utility of this method, we have transformed the products into a variety of other useful enantioenriched compounds (Fig. 3a). For example, β-aryl ketone can be directly transformed in good yields without racemization into terminal olefin, secondary alcohol, aromatic compound, ester, and amide (products 63–67).

a Transformations into other useful families of enantioenriched compounds. b Applications to the synthesis of natural products and bioactive molecules. CBS Corey-Bakshi-Shibata reagent; DEG diethylene glycol, TsOH p-toluenesulfonic acid, m-CPBA m-chloroperoxybenzoic acid, LDA lithium diisopropylamide.
Next, we applied our catalytic asymmetric synthesis of β-aryl ketone to a variety of target molecules, starting from commercially available ketones (Fig. 3b). For example, compound 68, a potent FXR (farnesoid X receptor) agonist analog121, can be prepared in two steps from acetophenone, via an aldol condensation followed by deoxygenative reductive cross-coupling. Pseudosinin I (69), a sesquiterpenoid obtained from Pseudotsuga sinensis122, can be produced in two steps from diacetone alcohol. (+)-ar-Juvabione (70), generated earlier in four steps via an enantioselective Heck arylation, exhibits juvenile hormone properties123. Using our method, we can obtain β-aryl ketone 70 in two steps and 90% ee from commercially available building blocks. Another natural product, (-)-dihydro-ar-Turmerone (71), which was previously generated in six steps via an asymmetric Michael addition and Dauben oxidation process, can be synthesized in two steps via our approach124.
We have conducted preliminary mechanistic studies of this deoxygenative reductive cross-coupling. In 2022, Zhou and co-workers presented an elegant method for the enantioselective reductive arylation of α,β-unsaturated ketones using nickel catalyst, which provided a highly efficient approach to β-aryl ketones125. Mechanistic studies revealed that arylnickel(I) species inserted into enones through 1,4-addition. On the other hand, MacMillan’s research have proposed the formation of organic radicals derived from alkyl alcohols114. In light of these findings, we are curious about whether our reaction occurs through an enone intermediate or an organic radical species.
The reaction of (E)-1-phenylhept-2-en-1-one was examined as the substrate in combination with an aryl bromide, in the absence of both NHC and 2,6-di-tert-butylpyridine. Interestingly, the desired product was successfully obtained, albeit with a slightly lower yield but maintaining the same ee (71% yield, 92% ee; Fig. 4a, entry 1). The introduction of a protic solvent (H2O) was found to be crucial in Zhou’s reaction. When we switched the mixed solvent system to pure MTBE, the yield for the reaction of enone was much lower compared to that of alcohol (Fig. 4a, entry 2). This observation indicates the involvement of distinct mechanisms for these two reactions. GC analysis of the model reaction (Table 1) showed that the enone species was generated in 0.10 equivalent after the initial step of NHC-alcohol adduct preparation, and maintained ~0.10 equiv throughout the entire coupling process. Kinetic studies suggested that the reaction starting from the alcohol substrate was approximately twice as fast as that starting from the enone substrate (Fig. 4a, right graph). Considering the additional time required for the formation of enone from the NHC-alcohol adduct, the likelihood of the coupling reaction proceeding through the enone intermediate is diminished.

a Comparison of the model reaction with the reaction of enone. b Radical trapping experiments. c Further evidence for the radical process and out-of-cage nickel-carbon bond formation. TEMPO (2,2,6,6-tetramethylpiperidin-1-yl)oxyl.
An alternative way to differentiate between the two pathways is by observing whether an organic radical is produced. Our findings provide evidence in support of the direct deoxygenative radical pathway. For example, if 1 equivalent of TEMPO (2,2,6,6-tetramethylpiperidin-1-yl)oxyl is added to a coupling in progress, carbon–carbon bond formation essentially ceases. The addition of 2 equivalents of allylic sulfone or vinyl phosphonate to the model reaction leads to a racemic adduct A1 or A2 without the formation of β-aryl ketone product (Fig. 4b).
When the β-hydroxy ketone illustrated in eq 1 (Fig. 4c) was subjected to the standard coupling conditions, the uncyclized product U was generated with 14% yield and 90% ee, along with the formation of cyclized product C in 65% yield and 1.6:1.0 dr (both diastereomers are racemic). This dr value is essentially identical to that obtained in an n-Bu3SnH-mediated reductive cyclization of the corresponding β-bromo ketone (1.7:1.0; Fig. 4c, eq 2), which is consistent with organic free radical R serving as a common intermediate in both processes. It has been reported that 5-hexenyl radicals cyclize with a rate constant of ~105 s-1 126, while the rate constant for diffusion is typically greater than 108 s-1 127. Thus, the identification of cyclized product C in eq 1 implies that the organic radical persists long enough to leave the solvent cage. An increase in the U/C ratio was observed with increasing nickel catalyst concentration (Fig. 4c, graph below eq 1), which suggests out-of-cage radical coupling instead of in-cage radical coupling. In contrast, the corresponding α,β-unsaturated ketone exclusively furnishes uncyclized product U (64% yield, 90% ee; Fig. 4c, eq 3), which supports a distinct 1,4-addition pathway in the coupling reaction of enone. Taken together, these observations suggest that the deoxygenative reductive cross-coupling reaction predominantly proceeds via an organic radical intermediate rather than an enone intermediate (for a proposed mechanism, see Supplementary Fig. 12).
Discussion
We have developed a nickel-catalyzed enantioconvergent deoxygenative reductive cross-coupling of unactivated alkyl alcohol (β-hydroxy ketone) and aryl bromide in the presence of an NHC activating agent. This scalable method tolerates substituents of varying sizes on the alcohol, and displays good functional-group tolerance. This approach features the utilization of two readily available coupling partners: alkyl alcohols and aryl bromides, enabling efficient and modular access to enantioenriched β-aryl ketones including a variety of interesting target molecules. Additional efforts to apply earth-abundant metals to useful asymmetric coupling reactions are underway in our lab.
Methods
General procedure for enantioconvergent deoxygenative reductive cross-coupling of alkyl alcohol and aryl bromide (alkyl alcohol: aryl bromide = 1.6: 1.0)
In a nitrogen-filled glovebox, an oven-dried 4 mL vial that contained a stir bar was charged with NiBr2·DME (8.0 mg, 0.025 mmol, 5.0 mol%), (S)-L1 (6.5 mg, 0.030 mmol, 6.0 mol%), and Ir[dF(CF3)ppy]2(dtbbpy)PF6 (9.0 mg, 0.0075 mmol, 1.5 mol%). Anhydrous isopropanol (1.5 mL) was added, and the vial was capped with a PTFE septum cap. The mixture was stirred at room temperature for 30 min, leading to a laurel-green solution. In a nitrogen-filled glovebox, a separate oven-dried 4 mL vial was charged with the alkyl alcohol (0.80 mmol, 1.6 equiv), NHC (316.5 mg, 0.80 mmol, 1.6 equiv), and a stir bar. Methyl tert-butyl ether (3.5 mL) was added, and the mixture was stirred at room temperature for 5 min. Next, 2,6-bis(tert-butyl) pyridine (179.5 μL, 0.80 mmol, 1.6 equiv) was added dropwise, and the resulting solution was stirred at room temperature for another 30 min (a white solid precipitated during this time). The suspension was filtered to furnish a homogeneous solution. In a nitrogen-filled glovebox, an oven-dried 20 mL vial was charged with the aryl bromide (0.50 mmol, 1.0 equiv), quinuclidine (67 mg, 0.60 mmol, 1.2 equiv), and a stir bar. The catalyst solution and NHC-alcohol adduct solution were transferred via syringe to the 20 mL reaction vial, followed by the addition of 4-methylpyridine (75 μL, 0.75 mmol, 1.5 equiv). The vial was transferred out of the glovebox and placed in an EtOH cooling bath at 10 °C for 5 min. Then the reaction was irradiated with blue LEDs (455 nm, 30 W) and was stirred at 10 oC for 18 h. The reaction mixture was passed through a plug of silica gel, and the vial, the cap, and the silica gel were rinsed with EtOAc. The filtrate was concentrated, and the residue was purified by flash chromatography on silica gel.
Data availability
The experimental data and the characterization data for all the compounds generated in this study are provided in the Supplementary Information. Experimental details: general information, preparation of alkyl alcohols, catalytic enantioconvergent cross-couplings, effect of reaction parameters, cross-couplings of other alkyl alcohols, comparison between the stability of alcohol and bromide, applications, mechanistic experiments, assignments of absolute configuration, NMR spectra and determination of stereoselectivity (PDF). CCDC 2281455, 2281456, 2281457 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cam-bridge CB2 1EZ, UK; fax: +44 1223 336033. All other data are available from the corresponding author upon request.
References
Lovering, F., Bikker, J. & Humblet, C. Escape from flatland: increasing saturation as an approach to Improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Geist, E., Kirschning, A. & Schmidt, T. sp3-sp3 Coupling reactions in the synthesis of natural products and biologically active molecules. Nat. Prod. Rep. 31, 441–448 (2014).
Cherney, A. H., Kadunce, N. T. & Reisman, S. E. Enantioselective and enantiospecific transition-metal-catalyzed cross-coupling reactions of organometallic reagents to construct C−C bonds. Chem. Rev. 115, 9587–9652 (2015).
Iwasaki, T. & Kambe, N. Ni-catalyzed C−C couplings using alkyl electrophiles. Top. Curr. Chem. 374, 66 (2016).
Fu, G. C. Transition-metal catalysis of nucleophilic substitution reactions: a radical alternative to SN1 and SN2 processes. ACS Cent. Sci. 3, 692–700 (2017).
Choi, J. & Fu, G. C. Transition metal–catalyzed alkyl-alkyl bond formation: another dimension in cross-coupling chemistry. Science 356, eaaf7230 (2017).
Kaga, A. & Chiba, S. Engaging radicals in transition metal-catalyzed cross-coupling with alkyl electrophiles: recent advances. ACS Catal. 7, 4697–4706 (2017).
Cheng, X., Lu, H. & Lu, Z. Enantioselective benzylic C-H arylation via photoredox and nickel dual catalysis. Nat. Commun. 10, 3549 (2019).
Gandolfo, E., Tang, X., Raha Roy, S. & Melchiorre, P. Photochemical asymmetric nickel-catalyzed acyl cross-coupling. Angew. Chem. Int. Ed. 58, 16854–16858 (2019).
Shu, X., Huan, L., Huang, Q. & Huo, H. Direct enantioselective C(sp3)–H acylation for the synthesis of α-amino ketones. J. Am. Chem. Soc. 142, 19058–19064 (2020).
Guo, L. et al. General method for enantioselective three-component carboarylation of alkenes enabled by visible-light dual photoredox/nickel catalysis. J. Am. Chem. Soc. 142, 20390–20399 (2020).
Cheng, X., Li, T., Liu, Y. & Lu, Z. Stereo- and enantioselective benzylic C–H alkenylation via photoredox/nickel dual catalysis. ACS Catal. 11, 11059–11065 (2021).
Chen, J. & Zhu, S. Nickel-catalyzed multicomponent coupling: synthesis of α-chiral ketones by reductive hydrocarbonylation of alkenes. J. Am. Chem. Soc. 143, 14089–14096 (2021).
Shu, X., Zhong, D., Lin, Y., Qin, X. & Huo, H. Modular access to chiral α-(hetero)aryl amines via Ni/photoredox-catalyzed enantioselective cross-coupling. J. Am. Chem. Soc. 144, 8797–8806 (2022).
Jiang, X., Sheng, F.-T., Zhang, Y., Deng, G. & Zhu, S. Ligand relay catalysis enables asymmetric migratory reductive acylation of olefins or alkyl halides. J. Am. Chem. Soc. 144, 21448–21456 (2022).
Li, X. et al. Three-component enantioselective alkenylation of organophosphonates via nickel metallaphotoredox catalysis. Chem 9, 154–169 (2023).
Xu, S. et al. Enantioselective C(sp3)–H functionalization of oxacycles via photo-HAT/nickel dual catalysis. J. Am. Chem. Soc. 145, 5231–5241 (2023).
Du, X., Cheng-Sánchez, I. & Nevado, C. Dual nickel/photoredox-catalyzed asymmetric carbosulfonylation of alkenes. J. Am. Chem. Soc. 145, 12532–12540 (2023).
Choi, J., Martín-Gago, P. & Fu, G. C. Stereoconvergent arylations and alkenylations of unactivated alkyl electrophiles: catalytic enantioselective synthesis of secondary sulfonamides and sulfones. J. Am. Chem. Soc. 136, 12161–12165 (2014).
Liang, Y. & Fu, G. C. Stereoconvergent negishi arylations of racemic secondary alkyl electrophiles: differentiating between a CF3 and an alkyl group. J. Am. Chem. Soc. 137, 9523–9526 (2015).
Huang, W., Wan, X. & Shen, Q. Enantioselective construction of trifluoro methoxylated stereogenic centers by a nickel-catalyzed asymmetric Suzuki–Miyaura coupling of secondary benzyl bromides. Angew. Chem. Int. Ed. 56, 11986–11989 (2017).
Varenikov, A. & Gandelman, M. Organotitanium nucleophiles in asymmetric cross-coupling reaction: stereoconvergent synthesis of chiral α‑CF3 thioethers. J. Am. Chem. Soc. 141, 10994–10999 (2019).
Everson, D. A., Shrestha, R. & Weix, D. J. Nickel-catalyzed reductive cross-coupling of aryl halides with alkyl halides. J. Am. Chem. Soc. 132, 920–921 (2010).
Zhao, Y. & Weix, D. J. Nickel-catalyzed regiodivergent opening of epoxides with aryl halides: co-catalysis controls regioselectivity. J. Am. Chem. Soc. 136, 48–51 (2014).
Zhao, Y. & Weix, D. J. Enantioselective cross-coupling of meso-epoxides with aryl halides. J. Am. Chem. Soc. 137, 3237–3240 (2015).
Huihui, K. M. M. et al. Decarboxylative cross-electrophile coupling of N-hydroxyphthalimide esters with aryl iodides. J. Am. Chem. Soc. 138, 5016–5019 (2016).
Kim, S., Goldfogel, M. J., Gilbert, M. M. & Weix, D. J. Nickel-catalyzed cross-electrophile coupling of aryl chlorides with primary alkyl chlorides. J. Am. Chem. Soc. 142, 9902–9907 (2020).
Franke, M. C. et al. Zinc-free, scalable reductive cross-electrophile coupling driven by electrochemistry in an undivided cell. ACS Catal. 12, 12617–12626 (2022).
Mouat, J. M. et al. CdS quantum dots for metallaphotoredox-enabled cross-electrophile coupling of aryl halides with alkyl halides. ACS Catal. 13, 9018–9024 (2023).
Cherney, A. H., Kadunce, N. T. & Reisman, S. E. Catalytic asymmetric reductive acyl cross-coupling: synthesis of enantioenriched acyclic α,α-disubstituted ketones. J. Am. Chem. Soc. 135, 7442–7445 (2013).
Cherney, A. H. & Reisman, S. E. Nickel-catalyzed asymmetric reductive cross-coupling between vinyl and benzyl electrophiles. J. Am. Chem. Soc. 136, 14365–14368 (2014).
Kadunce, N. T. & Reisman, S. E. Nickel-catalyzed asymmetric reductive cross-coupling between heteroaryl iodides and α‑chloronitriles. J. Am. Chem. Soc. 137, 10480–10483 (2015).
Suzuki, N., Hofstra, J. L., Poremba, K. E. & Reisman, S. E. Nickel-catalyzed enantioselective cross-coupling of N-hydroxyphthalimide esters with vinyl bromides. Org. Lett. 19, 2150–2153 (2017).
Poremba, K. E., Kadunce, N. T., Suzuki, N., Cherney, A. H. & Reisman, S. E. Nickel-catalyzed asymmetric reductive cross-coupling to access 1,1-diarylalkanes. J. Am. Chem. Soc. 139, 5684–5687 (2017).
Hofstra, J. L., Cherney, A. H., Ordner, C. M. & Reisman, S. E. Synthesis of enantioenriched allylic silanes via nickel-catalyzed reductive cross-coupling. J. Am. Chem. Soc. 140, 139–142 (2018).
DeLano, T. J. & Reisman, S. E. Enantioselective electroreductive coupling of alkenyl and benzyl halides via nickel catalysis. ACS Catal. 9, 6751–6754 (2019).
DeLano, T. J. et al. Nickel-catalyzed asymmetric reductive cross-coupling of α-chloroesters with (hetero)aryl iodides. Chem. Sci. 12, 7758–7762 (2021).
Turro, R. F., Brandstätter, M. & Reisman, S. E. Nickel-catalyzed reductive alkylation of heteroaryl imines. Angew. Chem. Int. Ed. 61, e202207597 (2022).
Lacker, C. R. et al. Enantioselective synthesis of N-benzylic heterocycles by Ni/photoredox dual catalysis. J. Am. Chem. Soc. 144, 20190–20195 (2022).
Turro, R. F. et al. Mechanistic investigation of Ni-catalyzed reductive cross-coupling of alkenyl and benzyl electrophiles. J. Am. Chem. Soc. 145, 14705–14715 (2023).
Woods, B. P., Orlandi, M., Huang, C.-Y., Sigman, M. S. & Doyle, A. G. Nickel-catalyzed enantioselective reductive cross-coupling of styrenyl aziridines. J. Am. Chem. Soc. 139, 5688–5691 (2017).
Banerjee, A. & Yamamoto, H. Nickel catalyzed regio-, diastereo-, and enantioselective cross-coupling of 3,4-epoxyalcohol with aryl Iodides. Org. Lett. 19, 4363–4366 (2017).
Guan, H., Zhang, Q., Walsh, P. J. & Mao, J. Nickel/photoredox-catalyzed asymmetric reductive cross-coupling of racemic α-chloro esters with aryl iodides. Angew. Chem. Int. Ed. 59, 5172–5177 (2020).
Zheng, P. et al. Dual Ni/photoredox-catalyzed asymmetric cross-coupling to access chiral benzylic boronic esters. Nat. Commun. 12, 1646 (2021).
Sun, D., Ma, G., Zhao, X., Lei, C. & Gong, H. Nickel-catalyzed asymmetric reductive arylation of α-chlorosulfones with aryl halides. Chem. Sci. 12, 5253–5258 (2021).
Min, Y. et al. Diverse synthesis of chiral trifluoromethylated alkanes via nickel-catalyzed asymmetric reductive cross-coupling fluoroalkylation. Angew. Chem. Int. Ed. 60, 9947–9952 (2021).
Lau, S. H. et al. Ni/photoredox-catalyzed enantioselective cross-electrophile coupling of styrene oxides with aryl iodides. J. Am. Chem. Soc. 143, 15873–15881 (2021).
Wang, D. & Xu, T. A pivotal role of chloride ion on nickel-catalyzed enantioselective reductive cross-coupling to perfluoroalkylated boronate esters. ACS Catal. 11, 12469–12475 (2021).
Wang, H., Zheng, P., Wu, X., Li, Y. & Xu, T. Modular and facile access to chiral α-aryl phosphates via dual nickel- and photoredox-catalyzed reductive cross-coupling. J. Am. Chem. Soc. 144, 3989–3997 (2022).
Wu, B.-B., Xu, J., Bian, K.-J., Gao, Q. & Wang, X.-S. Enantioselective synthesis of secondary β-trifluoromethyl alcohols via catalytic asymmetric reductive trifluoroalkylation and diastereoselective reduction. J. Am. Chem. Soc. 144, 6543–6550 (2022).
Jin, R.-X. et al. Asymmetric construction of allylic stereogenic carbon center featuring a trifluoromethyl group via enantioselective reductive fluoroalkylation. Nat. Commun. 13, 7035 (2022).
Li, T. et al. Enantioselective reductive cross-coupling of aryl/alkenyl bromides with benzylic chlorides via photoredox/biimidazoline nickel dual catalysis. Chin. J. Chem. 40, 1033–1038 (2022).
Zhou, J., Wang, D., Xu, W., Hu, Z. & Xu, T. Enantioselective C(sp3)-C(sp3) reductive cross-electrophile coupling of unactivated alkyl halides with α-chloroboronates via dual nickel/photoredox catalysis. J. Am. Chem. Soc. 145, 2081–2087 (2023).
Hu, X., Cheng-Sánchez, I., Cuesta-Galisteo, S. & Nevado, C. Nickel-catalyzed enantioselective electrochemical reductive cross-coupling of aryl aziridines with alkenyl bromides. J. Am. Chem. Soc. 145, 6270–6279 (2023).
Wang, H., Wu, X. & Xu, T. Enantioconvergent reductive C(sp)-C(sp3) cross-coupling to access chiral α-alkynyl phosphonates under dual nickel/photoredox catalysis. Angew. Chem. Int. Ed. 62, e202218299 (2023).
Wang, K., Ding, Z., Zhou, Z. & Kong, W. Ni-catalyzed enantioselective reductive diarylation of activated alkenes by domino cyclization/cross-coupling. J. Am. Chem. Soc. 140, 12364–12368 (2018).
Anthony, D., Lin, Q., Baudet, J. & Diao, T. Nickel-catalyzed asymmetric reductive diarylation of vinylarenes. Angew. Chem. Int. Ed. 58, 3198–3202 (2019).
Jin, Y. & Wang, C. Nickel-catalyzed asymmetric reductive arylalkylation of unactivated alkenes. Angew. Chem. Int. Ed. 58, 6722–6726 (2019).
Tian, Z.-X. et al. Highly Enantioselective cross-electrophile aryl-alkenylation of unactivated alkenes. J. Am. Chem. Soc. 141, 7637–7643 (2019).
He, J. et al. Nickel-catalyzed asymmetric reductive 1,2-carboamination of unactivated alkenes. Angew. Chem. Int. Ed. 59, 2328–2332 (2020).
Tu, H.-Y. et al. Enantioselective three-component fluoroalkylarylation of unactivated olefins through nickel-catalyzed cross-electrophile coupling. J. Am. Chem. Soc. 142, 9604–9611 (2020).
Wei, X., Shu, W., García-Domínguez, A., Merino, E. & Nevado, C. Asymmetric Ni-catalyzed radical relayed reductive coupling. J. Am. Chem. Soc. 142, 13515–13522 (2020).
Wu, X., Qu, J. & Chen, Y. Quinim: a new ligand scaffold enables nickel-catalyzed enantioselective synthesis of α‑alkylated γ‑lactam. J. Am. Chem. Soc. 142, 15654–15660 (2020).
Wu, X. et al. Catalytic desymmetric dicarbofunctionalization of unactivated alkenes. Angew. Chem. Int. Ed. 61, e202111598 (2022).
Cheng, L., Lin, Q., Chen, Y. & Gong, H. Recent progress on transition-metal-mediated reductive C(sp3)-O bond radical addition and coupling reactions. Synthesis 54, 4426–4446 (2022).
Pang, X., Su, P.-F. & Shu, X.-Z. Reductive cross-coupling of unreactive electrophiles. Acc. Chem. Res. 55, 2491–2509 (2022).
Pang, X. & Shu, X.-Z. Reductive deoxygenative functionalization of alcohols by first-row transition metal catalysis. Chin. J. Chem. 41, 1637–1652 (2023).
Do, H.-Q., Chandrashekar, E. R. R. & Fu, G. C. Nickel/bis(oxazoline)-catalyzed asymmetric Negishi arylations of racemic secondary benzylic electrophiles to generate enantioenriched 1,1-diarylalkanes. J. Am. Chem. Soc. 135, 16288–16291 (2013).
Chen, H.-W. et al. Asymmetric deoxygenative cyanation of benzyl alcohols enabled by synergistic photoredox and copper catalysis. Chin. J. Chem. 38, 1671–1675 (2020).
Luo, Y.-R. Handbook of Bond Dissociation Energies in Organic Compounds 1st edn, Vol. 392, 205–208 (CRC Press: Boca Raton, FL, 2002).
Anka-Lufford, L. L., Prinsell, M. R. & Weix, D. J. Selective cross-coupling of organic halides with allylic acetates. J. Org. Chem. 77, 9989–10000 (2012).
Cui, X. et al. Nickel-catalyzed reductive allylation of aryl bromides with allylic acetate. Org. Biomol. Chem. 11, 3094–3097 (2013).
Molander, G. A., Traister, K. M. & O’Neill, B. T. Engaging nonaromatic, heterocyclic tosylates in reductive cross-coupling with aryl and heteroaryl bromides. J. Org. Chem. 80, 2907–2911 (2015).
Wang, J., Zhao, J. & Gong, H. Nickel-catalyzed methylation of aryl halides/tosylates with methyl tosylate. Chem. Commun. 53, 10180–10183 (2017).
Komeyama, K., Ohata, R., Kiguchi, S. & Osaka, I. Highly nucleophilic vitamin B12-assisted nickel-catalysed reductive coupling of aryl halides and non-activated alkyl tosylates. Chem. Commun. 53, 6401–6404 (2017).
Vara, B. A., Patel, N. R. & Molander, G. A. O-benzyl xanthate esters under Ni/photoredox dual catalysis: selective radical generation and Csp3−Csp2 cross-coupling. ACS Catal. 7, 3955–3959 (2017).
Ackerman, L. K. G., Anka-Lufford, L. L., Naodovic, M. & Weix, D. J. Cobalt co-catalysis for cross-electrophile coupling: diarylmethanes from benzyl mesylates and aryl halides. Chem. Sci. 6, 1115–1119 (2015).
Konev, M. O., Hanna, L. E. & Jarvo, E. R. Intra- and intermolecular nickel-catalyzed reductive cross-electrophile coupling reactions of benzylic esters with aryl halides. Angew. Chem. Int. Ed. 55, 6730–6733 (2016).
Tao, X., Chen, Y., Guo, J., Wang, X. & Gong, H. Preparation of α-amino acids via Ni-catalyzed reductive vinylation and arylation of α-pivaloyloxy glycine. Chem. Sci. 12, 220–226 (2021).
Zhang, X. & MacMillan, D. W. C. Alcohols as latent coupling fragments for metallaphotoredox catalysis: sp3−sp2 cross-coupling of cxalates with aryl halides. J. Am. Chem. Soc. 138, 13862–13865 (2016).
Yan, X.-B., Li, C.-L., Jin, W.-J., Guo, P. & Shu, X.-Z. Reductive coupling of benzyl oxalates with highly functionalized alkyl bromides by nickel catalysis. Chem. Sci. 9, 4529–4534 (2018).
Gao, M., Sun, D. & Gong, H. Ni-catalyzed reductive C−O bond arylation of oxalates derived from α-hydroxy esters with aryl halides. Org. Lett. 21, 1645–1648 (2019).
Arendt, K. M. & Doyle, A. G. Dialkyl ether formation by nickel-catalyzed cross-coupling of acetals and aryl iodides. Angew. Chem. Int. Ed. 54, 9876–9880 (2015).
Pan, Y., Gong, Y., Song, Y., Tong, W. & Gong, H. Deoxygenative cross-electrophile coupling of benzyl chloroformates with aryl iodides. Org. Biomol. Chem. 17, 4230–4233 (2019).
Kariofillis, S. K., Shields, B. J., Tekle-Smith, M. A., Zacuto, M. J. & Doyle, A. G. Nickel/photoredox-catalyzed methylation of (hetero)aryl chlorides using trimethyl orthoformate as a methyl radical source. J. Am. Chem. Soc. 142, 7683–7689 (2020).
Parasram, M., Shields, B. J., Ahmad, O., Knauber, T. & Doyle, A. G. Regioselective cross-electrophile coupling of epoxides and (hetero)aryl iodides via Ni/Ti/photoredox catalysis. ACS Catal. 10, 5821–5827 (2020).
Kariofillis, S. K. et al. Using data science to guide aryl bromide substrate scope analysis in a Ni/photoredox-catalyzed cross-coupling with acetals as alcohol-derived radical sources. J. Am. Chem. Soc. 144, 1045–1055 (2022).
Suga, T. & Ukaji, Y. Nickel-catalyzed cross-electrophile coupling between benzyl alcohols and aryl halides assisted by titanium co-reductant. Org. Lett. 20, 7846–7850 (2018).
Suga, T., Takahashi, Y. & Ukaji, Y. One-shot radical cross coupling between benzyl alcohols and alkenyl halides using Ni/Ti/Mn system. Adv. Synth. Catal. 362, 5622–5626 (2020).
Diéguez, H. R. et al. Weakening C-O bonds: Ti(III), a new reagent for alcohol deoxygenation and carbonyl coupling olefination. J. Am. Chem. Soc. 132, 254–259 (2010).
Zheng, X. et al. Umpolung of hemiaminals: titanocene-catalyzed dehydroxylative radical coupling reactions with activated alkenes. Angew. Chem. Int. Ed. 52, 3494–3498 (2013).
Suga, T., Shimazu, S. & Ukaji, Y. Low-valent titanium-mediated radical conjugate addition using benzyl alcohols as benzyl radical sources. Org. Lett. 20, 5389–5392 (2018).
Xie, H. et al. Radical dehydroxylative alkylation of tertiary alcohols by Ti catalysis. J. Am. Chem. Soc. 142, 16787–16794 (2020).
Sumiyama, K. et al. Use of isopropyl alcohol as a reductant for catalytic dehydoxylative dimerization of benzylic alcohols utilizing Ti–O bond photohomolysis. Eur. J. Org. Chem. 2021, 2474–2478 (2021).
Xie, H., Wang, S., Wang, Y., Guo, P. & Shu, X.-Z. Ti-catalyzed reductive dehydroxylative vinylation of tertiary alcohols. ACS Catal. 12, 1018–1023 (2022).
Suga, T., Takahashi, Y., Miki, C. & Ukaji, Y. Direct and unified access to carbon radicals from aliphatic alcohols by cost-efficient titanium-mediated homolytic C–OH bond cleavage. Angew. Chem. Int. Ed. 61, e202112533 (2022).
Suga, T., Takada, R., Shimazu, S., Sakata, M. & Ukaji, Y. Highly (E)-selective trisubstituted alkene synthesis by low-valent titanium-mediated homolytic cleavage of alcohol C–O bond. J. Org. Chem. 87, 7487–7493 (2022).
Lin, Q., Tong, W., Shu, X.-Z. & Chen, Y. Ti-catalyzed dehydroxylation of tertiary alcohols. Org. Lett. 24, 8459–8464 (2022).
Li, Z. et al. Electrochemically enabled, nickel-catalyzed dehydroxylative cross-coupling of alcohols with aryl halides. J. Am. Chem. Soc. 143, 3536–3543 (2021).
Li, Y. et al. Chemoselective and diastereoselective synthesis of C-aryl nucleoside analogues by nickel-catalyzed cross-coupling of furanosyl acetates with aryl iodides. Angew. Chem. Int. Ed. 61, e202110391 (2022).
Lin, Q., Ma, G. & Gong, H. Ni-catalyzed formal cross-electrophile coupling of alcohols with aryl halides. ACS Catal. 11, 14102–14109 (2021).
Chi, B. K. et al. In-situ bromination enables formal cross-electrophile coupling of alcohols with aryl and alkenyl halides. ACS Catal. 12, 580–586 (2022).
Jia, X.-G., Guo, P., Duan, J. & Shu, X.-Z. Dual nickel and lewis acid catalysis for cross-electrophile coupling: the allylation of aryl halides with allylic alcohols. Chem. Sci. 9, 640–645 (2018).
Guo, P. et al. Dynamic kinetic cross-electrophile arylation of benzyl alcohols by nickel catalysis. J. Am. Chem. Soc. 143, 513–523 (2021).
Ma, W.-Y. et al. Cobalt-catalyzed enantiospecific dynamic kinetic cross-electrophile vinylation of allylic alcohols with vinyl triflates. J. Am. Chem. Soc. 143, 15930–15935 (2021).
Pan, F.-F., Guo, P., Huang, X. & Shu, X.-Z. Synthesis of dibenzyls by nickel-catalyzed homocoupling of benzyl alcohols. Synthesis 53, 3094–3100 (2021).
Chen, Y.-G. et al. Regioselective Ni-catalyzed carboxylation of allylic and propargylic alcohols with carbon dioxide. Org. Lett. 19, 2969–2972 (2017).
van Gemmeren, M. et al. Switchable site-selective catalytic carboxylation of allylic alcohols with CO2. Angew. Chem. Int. Ed. 56, 6558–6562 (2017).
Chenniappan, V. K., Peck, D. & Rahaim, R. Nickel catalyzed deoxygenative cross-coupling of benzyl alcohols with aryl-bromides. Tetrahedron Lett. 61, 151729 (2020).
Tercenio, Q. D. & Alexanian, E. J. Stereospecific nickel-catalyzed reductive cross-coupling of alkyl tosylate and allyl alcohol alectrophiles. Org. Lett. 23, 7215–7219 (2021).
Wang, H. et al. Nickel-catalyzed reductive Csp2−Csp3 cross coupling using phosphonium salts. Org. Lett. 23, 8183–8188 (2021).
Yu, H. & Wang, Z.-X. Nickel-catalyzed cross-electrophile coupling of aryl chlorides with allylic alcohols. Org. Biomol. Chem. 19, 9723–9731 (2021).
Fan, Z., Chen, S., Zou, S. & Xi, C. Direct C–C bond formation of allylic alcohols with CO2 toward carboxylic acids by ahotoredox/nickel dual catalysis. ACS Catal. 12, 2781–2787 (2022).
Dong, Z. & MacMillan, D. W. C. Metallaphotoredox-enabled deoxygenative arylation of alcohols. Nature 598, 451–456 (2021).
Sakai, H. A. & MacMillan, D. W. C. Nontraditional fragment couplings of alcohols and carboxylic acids: C(sp3)−C(sp3) cross-coupling via radical sorting. J. Am. Chem. Soc. 144, 6185–6192 (2022).
Intermaggio, N. E., Millet, A., Davis, D. L. & MacMillan, D. W. C. Deoxytrifluoromethylation of alcohols. J. Am. Chem. Soc. 144, 11961–11968 (2022).
Wang, J. Z., Sakai, H. A. & MacMillan, D. W. C. Alcohols as alkylating agents: photoredox-catalyzed conjugate alkylation via in situ deoxygenation. Angew. Chem. Int. Ed. 61, e202207150 (2022).
Lyon, W. L. & MacMillan, D. W. C. Expedient access to underexplored chemical space: deoxygenative C(sp3)−C(sp3) cross-coupling. J. Am. Chem. Soc. 145, 7736–7742 (2023).
Gould, C. A., Pace, A. L. & MacMillan, D. W. C. Rapid and modular access to quaternary carbons from tertiary alcohols via bimolecular homolytic substitution. J. Am. Chem. Soc. 145, 16330–16336 (2023).
Zhao, W.-T. & Shu, W. Enantioselective Csp3-Csp3 formation by nickel-catalyzed enantioconvergent cross-electrophile alkyl-alkyl coupling of unactivated alkyl halides. Sci. Adv. 9, eadg9898 (2023).
Schuster, D. et al. Pharmacophore-based discovery of FXR agonists. Part I: model development and experimental validation. Bioorg. Med. Chem. 19, 7168–7180 (2011).
Huang, T. et al. Phytochemical and biological studies on rare and endangered plants endemic to China. Part XV. structurally diverse diterpenoids and sesquiterpenoids from the vulnerable conifer Pseudotsuga sinensis. Phytochemistry 169, 112184 (2020).
Werner, E. W., Mei, T.-S., Burckle, A. J. & Sigman, M. S. Enantioselective heck arylations of acyclic alkenyl alcohols using a redox-relay strategy. Science 338, 1455–1458 (2012).
Khatua, A., Pal, S., Das, M. K. & Bisai, V. Asymmetric total syntheses of (–)-ar-turmerone, (–)-dihydro-ar-turmerone, (–)-ar-dehydrocurcumene, and (–)-ar-himachalene via a key allylic oxidative rearrangement. Tetrahedron Lett. 73, 153105 (2021).
Zhang, L. et al. Nickel-catalyzed enantioselective reductive conjugate arylation and heteroarylation via an elementary mechanism of 1,4-addition. J. Am. Chem. Soc. 144, 20249–20257 (2022).
Beckwith, A. L. J. & Schiesser, C. H. Regio- and stereo-selectivity of alkenyl radical ring closure: a theoretical study. Tetrahedron 41, 3925–3941 (1985).
Anslyn, E. V. & Dougherty, D. A. Modern Physical Organic Chemistry. 155–157 (University Science Books, 2006).
Acknowledgements
We thank Prof. Gregory C. Fu (Caltech), Prof. Shu-Li You (SIOC), and Prof. Chao Zheng (SIOC) for helpful suggestions and proofreading. Support has been provided by the National Natural Science Foundation of China (Grant No. 22201216), the National Key Research & Development Program of China (Grant No. 2023YFA1508600), and the Fundamental Research Funds for the Central Universities (Grant No. 22120230252, 2023-3-YB-10).
Author information
Authors and Affiliations
Contributions
Z.-P.Y. conceived and directed the project. L.-L.Z. and Y.-Z.G. discovered and developed the reaction. L.-L.Z., Y.-Z.G. and S.-H.C. performed the experiments and collected the data. H.Y., S.-J.S. and Q.P. discussed the project with Z.-P.Y. Z.-P.Y. wrote the paper with contributions from all authors. L.-L.Z. and Y.-Z.G. contributed equally.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Takuya Suga and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, LL., Gao, YZ., Cai, SH. et al. Ni-catalyzed enantioconvergent deoxygenative reductive cross-coupling of unactivated alkyl alcohols and aryl bromides. Nat Commun 15, 2733 (2024). https://doi.org/10.1038/s41467-024-46713-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-46713-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
