
Abstract
While aqueous organic redox flow batteries (RFBs) represent potential solutions to large-scale grid storage, their electrolytes suffer from short lifetimes due to rapid degradation. We show how an understanding of these degradation processes can be used to dramatically improve performance, as illustrated here via a detailed study of the redox-active biomolecule, flavin mononucleotide (FMN), a molecule readily derived from vitamin B2. Via in-situ nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR) we identify FMN hydrolysis products and show that these give rise to the additional plateau seen during charging of an FMN-cyanoferrate battery. The redox reactions of the hydrolysis product are not reversible, but we demonstrate that capacity is still retained even after substantial hydrolysis, albeit with reduced voltaic efficiency, FMN acting as a redox mediator. Critically, we demonstrate that degradation is mitigated and battery efficiency is substantially improved by lowering the pH to 11. Furthermore, the addition of cheap electrolyte salts to tune the pH results in a dramatic increase in solubility (above 1 M), this systematic improvement of the flavin-based system bringing RFBs one step closer to commercial viability.
Similar content being viewed by others
Introduction
Redox flow batteries (RFBs) are a promising energy storage system for grid-level storage, where low-cost and scalability are essential1. To date, many different organic molecules including quinones2,3,4, viologens5,6, phenazines7,8, and alloxazines7,9 have been investigated as potentially cheap RFB active molecules. Although a few molecules have shown a good performance in alkaline solution (pH 14)7,8, most organic molecules considered for RFBs generally experience degradation, reducing cell lifetime1. In 2016, Orita et al. 10 reported an RFB comprising flavin mononucleotide (FMN3−) at pH 14 as the anolyte against a potassium hexacyanoferrate K4[FeII(CN)6] catholyte. The cell showed a capacity retention of 99% over the course of 100 cycles. Despite the encouraging capacity retention, an additional FMN reduction plateau appeared during charge, which was assigned to a dimerization process10. The process was not seen on discharge, leading to considerable cell hysteresis. Consequently, the resulting capacity retention and energy efficiency were not good enough for grid-scale storage systems, where even longer long-life times with minimal degradation and high coulombic efficiency are required. More recently, Nambafu et al. attached a 2,2,6,6- tetramethylpiperidinyl-N-oxyl (TEMPO) radical to FMN to form a bifunctional redox active material, which showed improved stability at neutral pH11. However, significant capacity loss was seen within 100 cycles, which was largely ascribed to degradation of the TEMPO functionality.
FMN is a commercially available, non-toxic biomolecule, readily derived from vitamin B2, motivating its further study in an RFB10. This molecule is also used in the food industry as an orange-red food color additive, utilized in Europe as E101a12; the sodium salt is commonly known as E106 and is found in foods for babies and young children as well as jelly, milk products, and sweet products12.
Here, we demonstrate a powerful strategy to study the degradation of FMN3− by coupling in situ NMR and EPR techniques4. We explain how degradation, which we show involves the hydrolysis of FMN3− rather than a dimerization process, leads to the additional charging plateau. We investigate the electrochemical behavior of hydrolyzed FMN3− with in situ NMR and demonstrate that FMN3− acts as a redox mediator, chemically reducing the hydrolyzed product, explaining the lack of an additional plateau on discharge yet good cycling behavior, despite degradation and poor reversibility of hydrolyzed FMN’s redox reactions. Lastly, we provide a strategy to avoid hydrolysis by lowering the pH. The battery performance is improved significantly and the FMN solubility is increased dramatically, by addition of a simple salt.
Results and discussion
Galvanostatic cycling of an FMN-based RFB
To assess the cycling performance of FMN, an RFB was assembled comprising of 60 mM FMN3− and 1 M KOH in D2O as the anolyte and 0.2 M K4[Fe(CN)6] with 0.05 M K3[Fe(CN)6] in 1 M KOH in D2O as the catholyte. The cycling data (Fig. 1b) shows a plateau at 1.02 Vcell (αred) and a second, sloping plateau or process starting at 1.71 Vcell (γred) during the first charge (reduction of FMN3−); the theoretical capacity of 3.22 Ah L−1 was achieved (here: 3.18 Ah L−1) before the second plateau, γred, was completed (4.62 Ah L−1). On discharge (oxidation of FMN5−) only a single sloping plateau at 1.09 Vcell (αox) was seen, and 2.79 Ah L−1 was recovered. On subsequent cycles, an additional charge plateau centered at 1.41 Vcell (βred) grew in prominence over 90 cycles, resulting in a steady drop in the voltaic efficiency (Fig. S9). The plateau γred decreased rapidly in capacity until the 15th cycle where it was no longer observed, after which the charge capacity stabilized at 2.80 Ah L−1. No new plateaus were observed during discharge.

a Two one-electron redox reaction mechanism of FMN3− at pH 14. The ion on the left shows the skeletal structure of FMN3− (with the alloxazine structural unit in blue). The fully deprotonated state that dominates at pH 14 is shown. The negative charge on the nitrogen in FMN3− is in resonance with the two carbonyl groups. b Galvanostatic cycling of an RFB between 0.5 and 2.0 Vcell for the full cell, for 91 cycles with 60 mM FMN3− in 1 M KOH/D2O as the anolyte (15 mL) and 0.2 M K4[Fe(CN)6] and 0.05 M K3[Fe(CN)6] in 1 M KOH/D2O as the catholyte (15 mL). A current density of ±10 mA cm−2 was used. The active area of the electrode was 5 cm2, giving a current of ±50 mA. The efficiencies are presented in Supplementary Note 2 and Fig. S12. The catholyte K4[Fe(CN)6]/ K3[Fe(CN)6], which operates with a single process at 0.5 V versus the standard hydrogen electrode (SHE)10, is in excess, and thus the different processes seen on charging of the full cell are assigned to those of the anolyte and are thus labeled, αred, βred and γred. Those on discharging are similarly labeled αox.
Plateaus αred and αox can be attributed to the reduction and oxidation of FMN3−/5−, and oxidation and reduction of Fe(CN)64−/3−, respectively (Fig. 1a), no distinct plateaus for the separate one-electron reactions of FMN3−/5− being seen. Plateau γred is tentatively assigned to water reduction (HER), which is corroborated by the formation of gas bubbles (observed in the tubing, Fig. S15). Deuterium evolution has been shown to commence at 1.20 Vcell vs. [Fe(CN)6]3−/4− in a 2,6−dihydroxyanthraquinone/K4[Fe(CN)6] study3 and the literature shows that the thermodynamic potential for hydrogen evolution from water at pH 14 is around −1.2 V vs. SHE3,4. In this case, the water splitting commenced at higher voltages (1.71 Vcell vs. [Fe(CN)6]3−/4−) and we hypothesize that the hydrogen evolution is kinetically inhibited due to the carbon electrodes used in this work. The rapid decrease in this reaction with cycling is tentatively ascribed to a passivating film forming on the carbon paper used as the electrode. The reaction can be completely avoided by using a lower upper cut-off voltage of 1.7 V (see later).
The gradual appearance of plateau βred suggests that a new reduction process is occurring, that is distinct from the reduction of FMN3−, or the semiquinone FMN4−•, to FMN5−. This process was seen in the earlier work of Orita et al.10 where they hypothesized that the two charging plateaus result from reduction of the monomer and dimer of FMN3−. They argued that due to the slower kinetics of the dimer oxidation, compared to that of the monomer, the discharge curve does not mirror the charge curve. We, however, propose that this process is associated with a degradation product of FMN3−. To identify the species giving rise to the plateaus observed in the cycling data, a combination of in situ NMR and EPR measurements were performed.
In situ NMR/EPR studies of FMN as an anolyte
The redox reactions and degradation of FMN3− during battery operation were followed by NMR/EPR spectroscopy (Supplementary Note 3, Fig. S13). Two different samples were studied: a freshly prepared (“fresh sample”) and a four-day-old (“aged sample”) FMN3− solution.
The voltage profile (Fig. 2c) of a full cell containing freshly prepared FMN3− (60 mM in 1 M KOH/D2O) anolyte and corresponding in situ 1H NMR and EPR spectra are shown in Fig. 2d and e, respectively. At OCV (0.51 Vcell), the FMN3− ring protons, H9 and H6, were clearly seen in the in situ 1H NMR spectra at 7.25 ppm and 7.33 ppm, respectively, the shifts correlating well to those obtained via ex situ NMR 1- and 2-dimensional NMR measurements (Supplementary Note 1.2 and 1.3, Fig. S7-9). No evidence for any radicals was seen by EPR spectroscopy. Upon charge (at start of the plateau αred), the alloxazine ring proton signals (H6, H9) in the 1H NMR spectra disappeared immediately (Fig. 2d), while a resonance (at 337.8 mT) grew in the EPR spectra (Fig. 2e), indicating the formation of a radical. The formation of radical species correlated with the disappearance of the 1H NMR signals at the beginning of the αred plateau, the radical being assigned to FMN4−•, as shown with ex situ EPR measurements and DFT calculations (Supplementary Note 1.1, Figs. S1–6, 16). A reliable estimation of the radical concentration could not be obtained from this data, due to the low signal-to-noise of the EPR signal of FMN4−•, indicating that it was present in very low concentrations throughout cycling ([FMN4−•] < 5%; Fig. S3).

a Labeling of aromatic proton positions of FMN3− and FMN5−. b Voltage profile of a cell comprising freshly prepared 60 mM FMN3− versus 0.2 M K4[Fe(CN)6] and 0.05 M K3[Fe(CN)6] in 1 M KOH/D2O. A constant current density of ±10 mA cm−2 (±50 mA) was applied during cycling between 0.5 and 2.0 Vcell, with the 1H NMR (aromatic region) spectrum of the fresh 60 mM FMN3− solution before cycling (1). c 1H NMR and d EPR spectra of the same cell (full NMR spectra in Fig. S14). e Voltage profile of a cell that used an aged (4 days) 60 mM FMN3− electrolyte with the 1H NMR spectra of the aged solution before cycling (2) and during αred (3). f 1H NMR and g EPR spectra (position of the slices (2) and (3) are marked on f; full NMR spectra in Fig. S17, 18).
At the start of plateau γred, two new 1H NMR signals at 6.12 ppm and 6.34 ppm appeared, which are assigned to the aromatic protons on FMN5− (H6”, H9”). The EPR signal disappeared, indicating that all of the radicals have been reduced. No signals for FMN3− were seen, confirming the complete reduction of FMN3− to FMN5− during plateau αred. The reverse of the trends during the αred plateau was observed in both the NMR and EPR spectra during plateau αox, the 1H NMR signals (H6”, H9”) disappearing due to the formation of FMN4−•, as confirmed by the appearance of a signal (at 337.8 mT) in the EPR spectra. Note that the signals H6 and H9 did not appear again at the end of αox since the cell was not held long enough at low voltages to remove all the radicals, the data shown here being part of a longer cycling study (with full cycling shown in Fig. S14). The in situ 31P NMR spectra show a similar trend of disappearing and reappearing signals (Fig. S19).
The voltage profile (Fig. 2g) of a full cell containing the aged sample surprisingly contained the higher voltage βred plateau that was seen only after extended cycling of a fresh sample. Furthermore, the 1H NMR spectrum of the aged sample showed signs of FMN3− degradation containing a new pair of 1H NMR signals at 7.15 ppm and 7.28 ppm (H6*, H9*; Fig. 2f-II), shifted by ~0.07 ppm from the FMN3− resonances, in addition to those for FMN3− (Fig. 2b-I). On charging the full cell, and similar to the fresh sample, the 1H NMR signals (Fig. 2h) for FMN3− disappeared immediately and the EPR signal at 337.8 mT (Fig. 2i) due to FMN4−• was observed (at start of the plateau αred). The H6* and H9* 1H NMR signals, by contrast, remained (Fig. 2f-III), but gradually shifted towards higher frequencies in part due to the change in the bulk magnetic susceptibility caused by the FMN4−• radicals generated during plateau αred.
At the end of the αred plateau, the H6” and H9” 1H NMR signals for FMN5− appeared immediately, as was seen for the fresh sample. Not until plateau βred, however, did the signals H6* and H9* slowly disappear, their loss correlating with the gradual appearance of a new set of signals at 6.47 ppm and 6.83 ppm (H6*”, H9*”). That no EPR signals were seen during plateau βred, indicates that the reduction of the species giving rise to H6* and H9* does not generate any (EPR-observable) radicals. As before, no EPR signal was observed during γred and the intensity of the 1H NMR signals (H6”, H9”; H6*”, H9*”) remained constant.
On discharging and during plateau αox, the 1H NMR signals for FMN5− disappeared immediately. The signals for H6*” and H9*” disappeared more slowly, their intensity loss again correlating inversely with the appearance of the signals H6* and H9*. The EPR signal for FMN4−• (337.8 mT) was observed again. The in situ 31P NMR spectra also show a similar trend of disappearing signals (Fig. S20). Thus, based on the in situ NMR and EPR data, a second species was formed during aging (or by extended cycling) that is closely related to, but exhibits different electrochemical behavior from, FMN3−.
Hydrolysis of FMN
To determine the composition of the second species, we considered various degradation mechanisms of FMN during aging. First, we explored the photo-reduction of FMN3− to FMN4−•13 as a possible degradation mechanism. Different concentrations of FMN4−• were observed based on different degrees of illumination of the sample: FMN4−• was generated within the first 30 min and was present in a higher concentration than found in a sample prepared in the dark, which exhibited, on the basis of its ex situ 1H NMR spectrum, little to no evidence of FMN4−• (Fig. S1). However, no evidence for FMN4−• was observed after 4 days, with the 1H NMR spectrum showing sharpened signals (Fig. S9). This indicates that the FMN4−• can been re-oxidized (Supplementary Note 1.1), presumably by dissolved oxygen, and hence was no longer present in the aged sample.
Secondly, FMN3− has been reported to undergo hydrolysis under basic conditions (pH > 12), a process that can be accelerated at high temperatures14 (Supplementary Note 4.1, Fig. S21, 22, 29). To verify whether hydrolysis was also the source of the species formed after aging, a fresh solution of FMN3− was heated to 90 °C for 2 h to ensure complete hydrolysis, after which it was characterized by NMR spectroscopy. The one- and two-dimensional NMR spectra (Fig. S21–27) and Infrared spectroscopy (Fig. S28) were consistent with the formation of 4-(D-ribo-2,3,4-trihydroxypentyl-5’-phosphate)−3-oxo-3,4-dihydroquinoxaline-2-carboxylate, RQC3- (Fig. 3a), one of a number of hydrolysis products that have been proposed to form on hydrolysis of FMN3− under different conditions (Supplementary Note 4.2, Fig. S22)11,14,15.

a Alkaline hydrolysis of FMN3− to form RQC3− as the product. b 31P NMR spectra of (bottom-to-top) fresh, aged, and hydrolyzed 60 mM of FMN3− in 1 M KOH/D2O. c Suggested two-electron redox reaction of RQC3− at pH 14. d Voltage profile of a 60 mM RQC3− solution versus 0.2 M K4[Fe(CN)6] and 0.05 M K3[Fe(CN)6] in 1 M KOH/D2O full cell as a function of time; a constant current density of ±10 mA cm−2 (±50 mA) was applied during cycling between 0.5 and 2.0 Vcell. e 1H NMR spectra of the anolyte (RQC3−/RQC5−). The EPR and the full 1H NMR spectra can be found in the supporting information (Figs. S31 and S32).
The ex situ 31P NMR spectrum (Fig. 3b) of the fresh sample was dominated by a signal at 4.74 ppm from to the phosphate group of FMN3−, with a minor signal being seen at 4.84 ppm. After hydrolysis, the FMN3− signal almost completely disappeared and a new signal at 4.79 ppm was observed, which we ascribe to RQC3−. The aged sample showed two, almost equally intense signals for FMN3− and RQC3−, confirming that the same hydrolysis product is indeed formed on aging. The minor signal seen at 4.84 ppm in the fresh sample suggests that small traces of RQC3− were formed shortly after dissolution of FMN3−, the slight change in shift of the NMR signal being ascribed to pH changes.
To explore the electrochemical performance of RQC3−, the hydrolyzed sample was cycled and characterized via 1H NMR (Fig. 3c–e). At OCV, the 1H NMR spectra (Fig. 3e) showed two signals for RQC3− at 7.20 ppm and 7.31 ppm, which have identical chemical shifts to the H6* and H9* signals observed in the aged sample. During charge, no plateau αred (1.04 Vcell) was observed, confirming that no FMN3− is present in the solution and instead a plateau, solely from the reduction of RQC3− (β*red) at 1.32 Vcell is now seen. In the corresponding 1H NMR spectra, the signals assigned to RQC3− (H6*, H9*) gradually decreased, eventually disappearing at the end of the β*red process, with the concomitant appearance of two peaks at 6.70 ppm and 6.95 ppm. The shifts of the new peaks matched those of H6*” and H9*” seen in the reduced aged sample. The process at 1.71 Vcell (γred) due to water reduction was again observed, and no change in the 1H NMR signals were observed during this plateau, consistent with this assignment. Of note, no EPR signals were observed during either of the two plateaus β*red and γred (Fig. S31, 32), nor did H6* and H9* disappear immediately on charging—an indication that no rapid electron transfer between diamagnetic and paramagnetic species occurs in this electrolyte mixture3. This further indicates that no radicals were formed during β*red. We, therefore, propose that RQC3− undergoes a direct two-electron reduction reaction to form RQC5− (Fig. 3c), resulting in the β*red plateau and new 1H NMR signals.
On applying a negative current, the voltage dropped down to 0.5 Vcell without any evidence of electrochemical processes occurring. This suggested that RQC3− cannot be reversibly cycled in the same manner as FMN3−. The cyclic voltammetry (CV) shown in the supporting information is consistent with this proposal (Fig. S30).
Further experiments revealed that RQC5− could be re-oxidized by either lowering the cell voltage to below 0.5 Vcell (i.e., raising the potential at the anolyte) or increasing the time spent at the end of discharge at 0.5 Vcell (i.e., a voltage hold; Figs. S33–36; Supplementary Note 4.6), indicating that the kinetics of oxidation are much slower than those for reduction. However, both experiments showed that the completely hydrolyzed FMN can be reversibly cycled if held long enough at lower voltages, as a small fraction of the capacity was regained. We tentatively hypothesize that the difficulty of the reverse reaction is due to the structural rearrangement required to oxidize RQC5− (Figs. S33–35; Supplementary Note 4.5).
Redox mediator properties of FMN3−
We propose that an alternative process occurs during discharge of the full cell that facilitates the re-oxidation of RQC5−. To determine whether the presence of FMN3− plays a key role in this, a solution of RQC3− (15 mL, 60 mM) was fully reduced to RQC5− (Fig. 4a, b, Step I), while acquiring in situ 1H NMR spectra. Note that an upper cut-off voltage of 1.7 Vcell was used to avoid water reduction. An equal amount of fresh FMN3− (15 mL, 60 mM) was then added (in Step II) and the solution was allowed to equilibrate for 5 h, the signals for RQC5− slowly disappearing concomitant with an increase in the signals for RQC3− in the 1H-NMR spectra. No signals for either FMN3− or FMN5− were seen throughout Step II, which is ascribed to the oxidation of RQC5− by FMN3−, forming FMN4−• and eventually FMN5−, and the consequent rapid electron transfer processes between the diamagnetic and paramagnetic FMNn− ions. The battery was then charged (Step III), and even though FMN3− had been added to the solution, plateau α (associated with the reduction of FMN3−) was not observed, and instead plateau βred (associated with the reduction of RQC3−) was seen. This is supported by the in situ 1H NMR spectra: the appearance of the signals for RQC5− was inversely correlated with the disappearance of the signals for RQC3−. Signals for FMN5− were observed at the start of plateau βred and their intensity remained unchanged (no signals for FMN3− could be detected), confirming that an almost complete reduction of FMN3− had already occurred before the start of plateau βred. This is consistent with the reduction of FMN3−to FMN5− during the 5-hour equilibration period (step II), and the oxidation of RQC5− to RQC3−; we propose that this occurs via an intermolecular process, in which FMN3− acts as the redox mediator serving to oxidize RQC5− chemically.

a Electrochemical and chemical processes that occur during the reaction steps, I–V: (I) Electrochemical reduction of RQC3− to RQC5−, β*red (II) addition of FMN3− and the chemical redox reaction involving FMN3− as a redox mediator to form RQC3− and FMN5− during rest, (III) electrochemical reduction of RQC3− (with FMN5− remaining in solution), (IV) electrochemical oxidation of FMN5−, and the subsequent chemical reaction involving the oxidized product FMN3- with RQC5- to form RQC3− and FMN5−, (V) electrochemical reduction of RQC3− and FMN3−. Black arrows indicate electrochemical processes and red ones, coupled chemical reactions. b Voltage (black) and current (red) profiles for hydrolyzed FMN3− versus 0.2 M K4[Fe(CN)6] and 0.05 M K3[Fe(CN)6] in 1 M KOH/D2O full cell as a function of time, while undergoing the V reaction steps shown in (I). Pink and blue shading in (a) and (b) representing charging and discharging respectively, while gray represents reactions that occur during the hold at OCV, FMN3− being introduced at the beginning of this step. c In-situ 1H NMR spectra of the anolyte (RQC3−/RQC5− and FMN3−/FMN5−) during steps I-V.
During discharge (step IV), the αox plateau was observed, but with a higher capacity (2.36 Ah L−1) than expected if only FMN5− was oxidized to FMN3− (1.61 Ah L−1; capacity calculated based on 30 mM FMN3− as a result of dilution with 15 mL RQC3− and the assumption that RQC3− does not contribute towards the discharge capacity). This indicates that another species must also be oxidized during plateau αox, again consistent with redox mediation mechanism: RQC5− is oxidized by FMN3− forming RQC3− and FMN5−, which can then be electrochemically re-oxidized to FMN3−. However, the expected capacity of FMN3− and RQC3− combined (3.22 Ah L−1) was not observed suggesting that the redox mediator reactions are slower than the electrochemical oxidation of FMN5−. This is corroborated by the in situ 1H NMR spectra during step IV, as residual signals for RQC5−, in addition to those for FMN3− and RQC3−, could be observed at the end of plateau αox. During the following charge (step V), plateaus αred and βred were now observed. The same capacity was obtained as during the previous discharge cycle (step IV).
Additional experiments (Supplementary Note 5.1) probing the redox mediating effect of FMN3−, i.e., adding a shorter rest period (Fig. S40), an extra voltage holds (Fig S41), or less FMN3—containing electrolyte solution (Figs. S42, 43), confirm the mediating effect.
Avoiding hydrolysis of FMN3−
Since the hydrolysis of FMN3− occurs at a high OH− concentration (pH > 12) and is accelerated by higher temperatures, the pH of the electrolyte solutions was lowered to minimize hydrolysis and increase capacity retention. Supplementary note 6 provides a detailed discussion about the pH dependence and solubility of FMN (Fig. S44) and its electrochemistry at different pHs (galvanostatic cycling and CV) with and without buffers (Figs. S10, 11, 44–59). These experiments revealed that a pH of close to 11 represents the best compromise between degradation at high pH and solubility at low pH. Therefore, an anolyte solution was prepared from 60 mM FMN3− and 62 mM KOH in D2O to obtain a pH of 11.4. 1 M potassium chloride (KCl) was added to compensate for the reduced salt concentration (previously 1 M KOH was used). 62 mM KOH was added to the catholyte (K4[Fe(CN)6]/K3[Fe(CN)6] in 1 M KCl in D2O) to match the salt concentration to that in the anolyte. The RFB was cycled over a narrower voltage range (0.5–1.4 Vcell) to avoid the water splitting reaction. Over the course of 98 cycles, the electrochemical data (Fig. 5) only showed plateaus α†red and α†ox, which are associated with the redox reaction of FMN3− (Fig. 1a). No additional plateaus were observed during the initial charge cycles (unlike for the pH 14 sample, Fig. 1b). This confirms that lowering the pH below 12 prevents significant hydrolysis of FMN3−, leading to a noticeable improvement in capacity retention: the lifetime of this battery has been improved and the hydrolysis of FMN3− was successfully avoided over the course of battery cycling along with better efficiencies (Supplementary Note 7.3). We did not observe the increase in pH seen by Nambafu et al. during cycling of their bifunctional FMN derivative, which in their case led to degradation over the course of 100 cycles11. Furthermore, no evidence of alloxazine hydrolysis was observed in our studies. The small loss of capacity is ascribed to crossover of the active species (Supplementary Note 7.4, Figs. S63, 64).

Galvanostatic cycling of an RFB at pH 11.4 between 0.5 and 1.4 Vcell for 98 cycles (from cycle 2) with 0.2 M K4[Fe(CN)6] and 0.05 M K3[Fe(CN)6] in 1 M KCl/D2O as the catholyte (10 mL) and 60 mM FMN3− in 1 M KCl/D2O as the anolyte (10 mL). A current density of ±10 mA cm−2 (±50 mA) was used over 98 cycles (full cycling performance and efficiencies in Figs. S60, 61). Two stacked membranes were used to minimize cross-over. Cycling is shown from the second cycle, the 1st cycle showing additional capacity that arises from the trace amounts of oxygen present in the cell (Supplementary note 7.2, Fig. S62).
This work shows that the observed asymmetry in the galvanostatic profile of this battery system (and the resulting voltaic efficiency) can be explained by the presence of hydrolyzed FMN3−. No evidence, i.e., a second charging plateau, was seen for significant dimerization of FMN3−, which was previously ascribed to be the source of the second charge plateau10. We note that while dimerization has been seen in oxidized flavins16, dimerization is much less likely at higher pH due to the increased charge of the flavins and increased coulomb repulsion between these ions, which increases further on reduction. For example, since the fully reduced ion has five negative charges (FMN5−), dimerization would involve a species with ten negative charges.
Improving the solubility of FMN3−
FMN has multiple protonation states, and its solubility is likely to be extremely pH dependent. Thus, the solubility of FMN and the effect of salt concentration was then re-evaluated at a lower pH (Supplementary Note 5, Fig S44). The solubility limit of FMN3− at pH 14 (1 M KOH) was only 0.1 M10, but at pH 11.4, the addition of 1 M KCl to a solution of 62 mM KOH led to a much-improved FMN3− solubility of 1.8 M (Fig. S65). This solubility is even higher than achieved at pH 14 with the addition of the solubilizing agent nicotinamide, where it was possible to increase the solubility of FMN3− to 1.5 M. This brings the anolyte into the solubility requirements (1.0–2.0 M) required for a commercial aqueous organic RFB17. Furthermore, these results suggest that other simple salts may similarly help achieve higher solubility in a wider range of electrolytes. Excellent cycling performance was maintained at the lower pH of pH 11 with a higher concentration of 240 mM FMN3− and a higher current density of ±50 mA cm−2 as shown in Fig. S66.
Summary
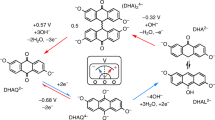
The use of online EPR/NMR metrologies to study degradation in an FMN (flavin mononucleotide)-based RFB is demonstrated in this work. Critically, this insight is then used to propose solutions to mitigate it. Via in- and ex-situ NMR spectroscopy, FMN3− in strongly alkaline solution is shown to be chemically unstable, forming the hydrolysis product RQC3−, either on aging the alkaline solution over the course of four days, or on electrochemical cycling. RQC3− is itself electrochemically active as shown by a new plateau at 1.42 Vcell (vs. Fe(CN)63−/ Fe(CN)64−) observed on charging solutions containing the hydrolysis products, in addition to the expected FMN reduction process at 1.02 Vcell. The new plateau, assigned to the two-electron reduction of RQC3− to form RQC5−, was not observed on discharge— unless deep discharging was used (i.e., a large overpotential was applied), indicating poor reversibility of the redox reaction. Despite this poor reversibility, solutions containing the hydrolysis products showed very little decrease in reversible capacity on galvanostatic cycling. Via a thorough assessment of the intermolecular processes using both NMR and EPR spectroscopy, we showed that the ability of FMN3− to act as a redox mediator was responsible for this phenomenon, and in solutions containing both FMN3− and its hydrolysis products, RQC5− is chemically re-oxidized to reform RQC3− (Fig. 6). While redox mediator processes are well established in for example lithium-air batteries18, their use in RFBs has been little discussed and may potentially offer new strategies for increase rates and reversibility.

a Reduction of FMN3−. b Oxidation of FMN5−. c Mediated electrochemistry of RQC5− and FMN3−.
In order to reduce the hysteresis in the electrochemical reaction, which reduces the overall voltaic efficiency of the reaction, we then explored approaches to reduce hydrolysis. To this end, lowering the pH to below pH 12 was found to prevent significant hydrolysis of FMN3−, leading to a noticeable improvement in capacity retention (Fig. 5) and both coulombic and voltaic efficiency. While, solubility is generally thought to lower the solubility of flavins, surprisingly, we found that the addition of KCl, required to maintain the salt concentration, was found to significantly improve the solubility, increasing from 0.1 M to 1.8 M on addition of KCl, leading to a higher solubility than observed in previous studies using the solubilizing agent nicotinamide10.
While any new chemistry requires detailed safety testing, this anolyte represents, one of the safest electrolytes/redox-active molecule currently available for flow batteries: FMN is a commercially available, non-toxic biomolecule widely used in the food industry. Moreover, the system works best at a lower pH, which also reduces the potential for corrosion, further enhancing the safety and easing the engineering requirements of the technology.
Methods
Materials
Flavin mononucleotide (FMN, 79% purity), potassium hexacyanoferrate(II) trihydrate (P3289, ≥98.5% purity), potassium hexacyanoferrate(III) (1049730100, ≥99.0%), ≥99.0%), and D2O (151882, 99.9 atom %) were purchased from Sigma Aldrich Chemicals. Potassium hydroxide (1917586, 86.4%) and potassium chloride (1858937, ≥99%) were purchased from Fisher Chemical. The aging process was conducted under air. The hydrolyzed FMN/RQC was synthesized by dissolving FMN in 1 M KOH/D2O and heating for 2 h at 90 °C. The solutions with a specific pH were made by dissolving a specific amount of FMN in 1 M KCl/D2O; the pH was then adjusted by the addition of KOH.
Flow battery assembly
The hardware of the flow battery was purchased from Scribner Associates. Graphite flow plates with serpentine flow patterns were used for both electrodes. Each electrode comprised of three pieces of Sigracet 39 AA carbon paper (FuelCellStore) with 5 cm2 active area, which were used without further treatment. Nafion 212 was used as the ion transport membranes. Pre-treatment of the Nafion 212 membranes was performed by first heating the membrane in 80 °C deionized water for 20 min and then soaking it in 5% hydrogen peroxide solution for 35 min. All battery performance experiments were carried out using a potentiostat (SP-150, BioLogic SAS)3,4.
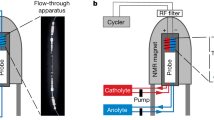
Online EPR and NMR setup
The setup consists of a flow battery (Scribner), two peristaltic pumps (MasterFlex L/S 07751-20, ColeParmer), an electrochemical cycler (SP-150, BioLogic SAS), a benchtop EPR (MS5000, Magnettech), and an NMR (300 MHz, Bruker) spectrometer. The battery and the EPR spectrometer were positioned outside the 5 G line of the NMR magnet. The electrolyte was pumped through the flow battery, then flowed through the EPR and NMR magnets, and finally back to the electrolyte reservoir. The direction of flow was from the bottom to the top of both magnets. PFA tubes (1/16 in.) were used to connect the electrolyte reservoir, the battery, and the EPR and NMR sampling tubes. It took 64 s, at a flow rate of 13.6 mL min−1, for the electrolyte to move through the system. From the electrolyte reservoir to the battery takes 3 s, from the battery to the EPR detection region 3 s, from the EPR to the NMR detection 29 s and from the NMR back to the electrolyte reservoir is 29 s. To minimize heating of the aqueous solution by microwave irradiation, a flat EPR cell (E4503, Magnettech, g = 2 at 336 Gauss) was used. A customized adaptor made of polyether ether ketone (PEEK) connected the flat EPR cell to the 1/16 in. tube. The cell was orientated in the resonator such that the strength of the magnetic field was maximized, and the strength of the electric field was minimized across the sample. The volume of the cell in the excitation region of the microwave was 0.03 mL (2.00 cm × 0.50 cm × 0.03 cm), giving a residence time of 0.13 s for the electrolyte solution at a flow rate of 13.6 mL min−1. Details of the NMR sampling tube were provided in our previous publication3,4.
For the coupled in situ EPR and NMR experiment, 30 mL of 60 mM FMN was used as the anolyte, 30 mL of 150 mM K4[Fe(CN)6] and 37.5 mM K3[Fe(CN)6] was used as the catholyte. The solvent was D2O, with 1 M KOH as the supporting electrolyte. The flow rate was 13.6 mL min−1.
Pseudo-2D NMR experiments were performed by direct excitation with a 90° radio-frequency pulse. Each NMR spectrum was acquired by collecting 16 free induction decays (FIDs) with a recycle delay of 5 s. The pulse length for a 90° pulse was 27 μs. All spectra were referenced to the chemical shift of water at 4.79 ppm before battery cycling began.
For the in situ EPR measurement of FMN4−• radical anions, the magnetic field was swept from 336.5 to 339 mT. The sweep time was 60 s per single scan. Bm was 0.001 mT, and Q was recorded for each spectrum. Q was 1306 ± 5. The temperature of the resonator was kept at 29 °C, and a time delay of 35 s was added between each scan.
Cyclic voltammetry
CV experiments were carried out using a three-electrode cell. A Thermo Haake DC50-K75 circulator was used to ensure that the voltametric cell was at 25 °C and GL14 screw threads and caps, in combination with rubber O-rings, were used to ensure a tight seal around the electrodes. A rubber septum was used to close the remaining opening, and for addition and removal of samples under inert atmosphere. The cell was flushed with nitrogen prior to measurement and kept under an overpressure of the inert gas during measurement using the ground-glass opening at the top of the cell. The nitrogen was dried over a desiccant column before use. All voltametric experiments used a glassy carbon working electrode embedded in a PEEK housing (Biologic A-012744, 3 mm) and a coiled platinum counter electrode (1.00 mm diameter). The glassy carbon was polished using 0.05 µm polishing alumina paste (Biologic A-001050) on alumina polishing pads (Biologic A-001040), and where necessary using 1 µm polishing diamond paste (Biologic A-002054) on diamond polishing pads (A-001041) with Millipore deionized water. 3 × 100 cycles of figure-eight were used to polish the working electrode, with intermediate sonication steps of one second duration followed by drying under nitrogen gas. The platinum was purchased from Sigma-Aldrich and cleaned using nitric acid and deionized water before use. A biologic VSP 200 potentiate was used, with the potentiostat software, for the experiments described here.
Electrolyte and supporting electrolyte solutions were prepared under inert atmosphere, avoiding the use of vacuum grease where possible, prior to measurement. A background CV was run on the supporting electrolyte solution to ensure that it was sufficiently free from contamination prior to use. 20 mVs−1 was chosen as the comparison scan-rate, providing a compromise between duration of the experiment and sufficient time to allow relaxation and separation of the different redox processes. Cyclic voltammetry was run over long time periods where possible to investigate the stability of the analyte against the supporting electrolyte fully, and to observe if alternative detrimental processes, such as electrode deposition, occurred.
Density functional theory calculations
The software package Gaussian 1619 was used for all calculations. The molecules were first optimized at the PBE20,21/TZVP22,23 level of theory without any solvation model applied. The optimized coordinates were then used as the initial structure for optimization at the PBE/TZVP level of theory with an applied polarized continuum model (PCM)24 of solvation using the dielectric constant of water. These optimized structures were then further optimized at the B3LYP25,26,27,28/TZVP level of theory, again using the PCM model for water. The energetics and vibrational frequencies were calculated for all species of interest. The absence of imaginary frequencies confirms that the obtained geometry corresponds to a ground state structure.
Data availability
Data underlying this paper is freely available at https://doi.org/10.17863/CAM.99832.
Code availability
All code used in this work is available from the corresponding author upon reasonable request.
References
Winsberg, J., Hagemann, T., Janoschka, T., Hager, M. D. & Schubert, U. S. Redox-flow batteries: from metals to organic redox-active. Mater. Angew. Chem. - Int. Ed. 56, 686–711 (2017).
Lin, K. et al. Alkaline quinone flow battery. Science 349, 1529–1532 (2015).
Zhao, E. W. et al. In situ NMR metrology reveals reaction mechanisms in redox flow batteries. Nature 579, 224–228 (2020).
Zhao, E. W. et al. Coupled in situ NMR and EPR studies reveal the electron transfer rate and electrolyte decomposition in redox flow batteries. J. Am. Chem. Soc. 143, 1885–1895 (2021).
Hu, B., DeBruler, C., Rhodes, Z. & Liu, T. L. Long-cycling aqueous organic redox flow battery (AORFB) toward sustainable and safe energy storage. J. Am. Chem. Soc. 139, 1207–1214 (2017).
Jin, S. et al. Near neutral pH redox flow battery with low permeability and long-lifetime phosphonated viologen active species. Adv. Energy Mater. 10, 1–10 (2020).
Lin, K. et al. A redox-flow battery with an alloxazine-based organic electrolyte. Nat. Energy 1, 1–8 (2016).
Hollas, A. et al. A biomimetic high-capacity phenazine-based anolyte for aqueous organic redox flow batteries. Nat. Energy 3, 508–514 (2018).
Wei, X. et al. Materials and systems for organic redox flow batteries: status and challenges. ACS Energy Lett. 2, 2187–2204 (2017).
Orita, A., Verde, M. G., Sakai, M. & Meng, Y. S. A biomimetic redox flow battery based on flavin mononucleotide. Nat. Commun. 7, 1–8 (2016).
Nambafu, G. S. et al. An organic bifunctional redox active material for symmetric aqueous redox flow battery. Nano Energy 89, 106422 (2021).
Turck, D. et al. Dietary reference values for riboflavin. EFSA J. 15, e04919 (2017).
Pompe, N., Illarionov, B., Fischer, M., Bacher, A. & Weber, S. Completing the picture: determination of 13 C hyperfine coupling constants of flavin semiquinone radicals by photochemically induced dynamic nuclear polarization spectroscopy. J. Phys. Chem. Lett. 13, 5160–5167 (2022).
Smith, S. B. & Bruice, T. C. Mechanisms of isoalloxazine (flavine) hydrolysis. J. Am. Chem. Soc. 97, 2875–2881 (1975).
Surrey, A. R. & Nachod, F. C. Alkaline hydrolysis of riboflavin. J. Am. Chem. Soc. 73, 2336–2338 (1951).
Bastian, M. & Sigel, H. The self-association of flavin mononucleotide (FMN2-) as determined by 1H NMR shift measurements. Biophys. Chem. 67, 27–34 (1997).
Darling, R. M., Gallagher, K. G., Kowalski, J. A., Ha, S. & Brushett, F. R. Pathways to low-cost electrochemical energy storage: a comparison of aqueous and nonaqueous flow batteries. Energy Environ. Sci. 7, 3459–3477 (2014).
Liu, T. et al. Cycling Li-O2 batteries via LiOH formation and decomposition. Science 350, 530–533 (2015).
Frisch, M. J. et al. Gaussian. (2016).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 78, 1396–1396 (1997).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Schäfer, A., Huber, C. & Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 100, 5829–5835 (1994).
Schäfer, A., Horn, H. & Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577 (1992).
Scalmani, G. & Frisch, M. J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 132, 114110 (2010).
Becke, A. D. Density-functional thermochemistry. III The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993).
Stephens, P. J., Devlin, F. J., Chabalowski, C. F. & Frisch, M. J. Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627 (1994).
Vosko, S. H., And, S. A., Ilk, L. W. & Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis Publication Date Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis 1. Can. J. Phys. 58, 1200 (1980).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785 (1988).
Acknowledgements
D.H. acknowledges support from the Sheldrick Scholarship in Chemistry, Jesus College Cambridge, and the EPSRC iCASE PhD Fees Only Studentship. C.P.G. acknowledges support from the European Research Council (ERC) 835073 BATNMR. D.H., R.J., E.W.Z., N.L.F., and C.P.G. acknowledge support from Shell; R.J. and N.L.F. acknowledge support from the EPSRC and Shell via I-Case studentships EP/R511870/1 and EP/V519662/1, respectively. R.B.J. thanks the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 101034413 for funding. E.W.Z. and C.P.G. acknowledge support from Centre of Advanced Materials for Integrated Energy Systems (CAM-IES), via EPSRC grant number EP/P007767/1. We thank D.S. Wright, A.C. Forse, M. De Volder, and E.J. Latchem from University of Cambridge and P.A.A. Klusener from Shell for many fruitful discussions.
Author information
Authors and Affiliations
Contributions
Conceptualization: D.H., E.W.Z., and C.P.G. Methodology: D.H., E.W.Z., R.B.J., N.L.F., and B.L.D.R. Investigation: D.H. Visualization: D.H. Project administration: C.P.G. Supervision: R.B.J., E.W.Z., and C.P.G. Writing—original draft: D.H. Writing—review and editing: R.B.J., B.L.D.R., and C.P.G.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hey, D., Jethwa, R.B., Farag, N.L. et al. Identifying and preventing degradation in flavin mononucleotide-based redox flow batteries via NMR and EPR spectroscopy. Nat Commun 14, 5207 (2023). https://doi.org/10.1038/s41467-023-40649-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-40649-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
