
Abstract
Studies of vacancy-mediated anomalous transport properties have flourished in diverse fields since these properties endow solid materials with fascinating photoelectric, ferroelectric, and spin-electric behaviors. Although phononic and electronic transport underpin the physical origin of thermoelectrics, vacancy has only played a stereotyped role as a scattering center. Here we reveal the multifunctionality of vacancy in tailoring the transport properties of an emerging thermoelectric material, defective n-type ZrNiBi. The phonon kinetic process is mediated in both propagating velocity and relaxation time: vacancy-induced local soft bonds lower the phonon velocity while acoustic-optical phonon coupling, anisotropic vibrations, and point-defect scattering induced by vacancy shorten the relaxation time. Consequently, defective ZrNiBi exhibits the lowest lattice thermal conductivity among the half-Heusler family. In addition, a vacancy-induced flat band features prominently in its electronic band structure, which is not only desirable for electron-sufficient thermoelectric materials but also interesting for driving other novel physical phenomena. Finally, better thermoelectric performance is established in a ZrNiBi-based compound. Our findings not only demonstrate a promising thermoelectric material but also promote the fascinating vacancy-mediated anomalous transport properties for multidisciplinary explorations.
Similar content being viewed by others

Introduction
Transport properties of a solid material can be mediated by structural defects that breed multifarious novel behaviors for applications in thermotics, electronics, optics, magnetics, etc1,2,3,4. Vacancy has been identified as an important type of defect and its versatility has been manifested in diverse areas, e.g., anomalous d0 ferromagnetism in dielectric oxides5,6, the delicate quantum nature of color centers in diamond7,8, and Rashba spin-orbit coupling in inversion-symmetry-breaking systems9. However, the functionality of vacancy seems sterile in thermoelectrics, where it basically acts as a phonon scattering center10,11. Recent investigations of vacancy-ordering-induced topological electronic transition in Eu2ZnSb212 and vacancy-related strain engineering in PbTe13 are particularly impressive for their high thermoelectric performance results, signifying that vacancy could play a more pivotal role in advancing thermoelectrics beyond its phonon scattering functionality.
The emerging 19-electron half-Heusler (HH) materials provide an ideal platform for exploration of vacancy-mediated anomalous transport properties since their inherent vacancy is vital to their structural stability14,15. In contrast to a typical 18-electron HH with unfilled anti-bonding states, a stoichiometric 19-electron HH is electron-sufficient and its densely occupied anti-bonding states will thus increase its instability. Following the valence-balanced rule, the defective structure of a stoichiometric 19-electron HH accommodating a large concentration of cationic vacancies is energetically preferred, and a number of stable compounds have also been experimentally reported16,17,18. A thorough examination of the thermodynamic stability and delicate control of vacancy concentration has been undertaken to discover new defective 19-electron HHs that extend the family of Heuslers19,20,21. Meanwhile, the major drawback of thermoelectric HHs—their intrinsically high lattice thermal conductivity (κlat)—has been addressed to some extent (Fig. 1)18,22,23. For instance, the room-temperature κlat of defective Nb0.8CoSb is ~4.2 W m−1 K−1, while that of NbCoSn is ~7.7 W m−1 K−1. There is no doubt that vacancy-mediated short phonon relaxation time will give rise to such reduced κlat. Nonetheless, the question whether vacancy endows defective 19-electron HHs with other anomalous transport properties remains nearly unaddressed. It is thus compelling to dig more deeply into the multifunctionality of vacancy in tailoring the phononic and electronic transport properties of defective 19-electron HHs.

Here we first describe an emerging defective ZrNiBi with a pure HH phase. Its randomly distributed zirconium vacancy is revealed using high-angle annular dark-field scanning transmission electron microscopy. Another distinctive structural characteristic is the existence of twofold twinning, which is rarely reported in HHs. More intriguingly, this material is distinguished among HHs due to its anomalous phononic and electronic transport mediated by its vacancy. An exceptionally low κlat of ~1.4 W m−1 K−1 at room temperature is obtained in defective ZrNiBi, the lowest among the HH family and comparable to that of other outstanding thermoelectric materials (Fig. 1). Besides the contribution of vacancy-mediated short phonon relaxation time, the phononic dynamics elucidates that the origin of such low κlat also correlates with vacancy-induced local soft bonds, low-lying optical phonons, and anisotropic lattice vibrations. In addition, the electronic band structure is modified by the cationic vacancy so that a flat band is observed near the conductive band minimum, which is unique among HHs and of great importance in physics. The flat band results in a large density of states near the band edge, which is particularly desirable for the electron-sufficient thermoelectric materials since large density of states effective mass requires high optimal carrier concentration. Finally, further carrier concentration optimization endows the defective ZrNiBi-based material with better thermoelectric properties, allowing it to outperform various typical n-type HHs. We believe that our findings of vacancy-mediated anomalous transport in defective ZrNiBi will stimulate the discovery of novel phenomena and the development of both innovative theories and potential technological applications.
Results
Crystallographic features
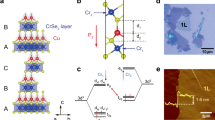
According to the Open Quantum Materials Database24, the stoichiometric ZrNiBi with a valence electron count (VEC) of 19 lies above the convex hull (hull distance is ~0.065 eV/atom) constructed by connecting the vertices based on the T = 0 K formation energies of all phases in its composition space, indicating that it is thermodynamically unstable. This is consistent with the X-ray diffraction (XRD) results shown in Fig. 2a, in which additional ZrNi alloy peaks can be observed. On the other hand, although Zr0.75NiBi is valence balanced [net valence, NV = 3 (0.75Zr4+ s0d0) − 0 (Ni0 d10) − 3 (Bi3− s2p6) = 0], Bi impurity phases were detected in this composition. By artificially tuning the Zr vacancy concentration to ~12% (i.e., Zr0.88NiBi), phase-pure samples can be repeatedly achieved. It should be noted that the XRD patterns for Zr0.87NiBi samples from two different batches showed contrasting phase purity despite the identical synthesis method used for each, where the sample from Batch I was found to be phase-pure while a tiny amount of Bi impurity was observed in the Batch II sample (Figure S1a). We speculate that the maximum Zr vacancy concentration for achieving stable phase purity is ~12%, and Zr0.88NiBi is thus designated for further discussion. Like typical 18-electron HHs, Zr0.88NiBi is crystallized in a cubic structure with space group \(F\bar{4}3m\), in which Bi (purple) and Ni (red) atoms form a tetrahedrally bonded sublattice with Zr (green) atoms partially filling the octahedral voids around the tetrahedral sublattice, as schematically illustrated in Fig. 2b (the white sphere represents Zr vacancy, VZr).

a VZr-concentration-dependent XRD patterns for ZrNiBi-based compounds. b Crystal structure of phase-pure Zr0.88NiBi. Green, red, purple, and white spheres represent Zr, Ni, Bi, and Zr vacancy, respectively. c Low-magnification TEM image of Zr0.88NiBi. Twinning-structure-induced string contrasts are indicated by arrows. d Atomic-resolution HAADF-STEM image showing the twofold twinning structure of Zr0.88NiBi. Yellow dash-dotted lines represent the boundary between the matrix and the primary twin T1 and blue dashed lines delineate the region of the secondary nanotwin T2. Inset: corresponding FFT diffraction pattern showing the superposition of diffraction spots from the matrix (red solid circles), the primary twin (yellow solid circles), and the secondary nanotwin (blue dotted circles). e HAADF-STEM image showing the atomic configuration of a single domain of Zr0.88NiBi and f the corresponding intensity profile within the 4a plane (region marked by the yellow dashed rectangle in e).
Moreover, a JEM-ARM200F Cs-corrected transmission electron microscope (TEM) was employed to reveal the microstructure of Zr0.88NiBi. As displayed in the low-magnification TEM image in Fig. 2c, visible string contrasts indicated by arrows are ubiquitous throughout the material, and these stem from the existence of a high density of twins. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image in Fig. 2d further illustrates the atomic configuration of this twinning structure. The shared twin plane between the matrix and the primary twin T1 is the \((\bar{1}11)\) plane and the twin angle is ~55°, which corresponds well with the theoretical angle of 54.7° between the \((\bar{1}11)\) and (002) planes. Interestingly, a secondary nanotwin T2 (delineated by blue dashed lines) is observed inside the primary twin T1. According to the atomistic dynamics of the self-activated hierarchical twinning behavior25, the emission of Shockley partial dislocations from the intrinsic kinks on the twin boundaries will occur when the kink height is greater than five atomic layers, which in turn induces the nucleation and growth of secondary nanotwins. The unique twinning structure is also verified by the corresponding fast Fourier transform (FFT) (inset, Fig. 2d), in which the superposition of diffraction spots from the matrix, the primary twin, and the secondary nanotwin (marked by different colors) is evident. With its twofold twin, Zr0.88NiBi is distinguished among the family of HHs since reports of twinned HHs have been rare. The contributing role of high-energy ball-milling and annealing processes in the formation of twinning structures has been discussed for various materials26,27, but it has not been reported for either ball-milled or annealed HHs. It should be noted that the “ball-milling + hot-pressing”-synthesized Zr0.88NiBi is not phase-pure, but the annihilation of the impurity phase can be observed following an additional annealing process (Figure S1b). Such recrystallization behavior could correlate with thermally activated atomic diffusion, as well as with twinning-process-associated atomic rearrangement28,29, where the formation of twins enables the existence of an interfacial network at a low energy state and thus stabilizes the structure30.
In addition to the unique twinning configuration, the structure also accommodates a large concentration of cationic vacancy. Figure 2e presents the HAADF-STEM image of a single domain taken along the [110] direction. Crystallographically, the specific atom on each Wyckoff position 4a (\(0,\,0,\,0\)) (i.e., Zr), 4b (\(\frac{1}{2},\,\frac{1}{2},\,\frac{1}{2}\)) (i.e., Bi), and 4c (\(\frac{1}{4},\,\frac{1}{4},\,\frac{1}{4}\)) (i.e., Ni) is visible without any overlapping along this direction. To verify the existence of Zr vacancies, the intensity profile within the 4a plane (region marked by a yellow dashed rectangle) was processed. As displayed in Fig. 2f, the intensity reduction detected at different positions corresponding to the orange solid circles within the yellow dashed rectangle in Fig. 2e indicates that the Zr sites are not fully occupied. In contrast, only a slight intensity fluctuation of Bi atoms can be observed (Figure S2). In addition, the intrinsic Zr vacancies seem to be randomly distributed rather than exhibiting short-range order (SRO), which was discovered in other defective 19-electron HHs (e.g., Nb0.8CoSb22). This can be ascertained from the dissimilar diffraction patterns for Nb0.8CoSb and Zr0.88NiBi, in which the presence and absence, respectively, of a SROed-vacancy-induced diffuse band is observed. Xia et al. found that the SRO configuration favors a lower energy state22, but the twinning configuration shows similar efficacy. In addition, stacking fault energy could be another coupling factor since SRO has a positive influence on it31, while twins are more easily formed in face-centered-cubic materials with low stacking fault energy30. As a result, competition between these two atomic configurations may occur. It should also be noted that the microstructure is highly sensitive to the synthesis process used, which allows for the possibility to finely control the atomic configuration, as well as the structure–property relationship, of defective HHs.
Phononic dynamics
It is foreseeable that the intrinsic vacancies and twins in Zr0.88NiBi will endow it with strong phonon scattering. According to the Klemens model32, point defects mainly scatter the high-frequency phonons and the scattering rate depends on the atomic mass fluctuation caused by the point defect. In principle, vacancy will produce much stronger fluctuation than substitutional impurity and thus acts as a more effective scattering source. Nonetheless, the phonon scattering effect of twinning structures remains a subject of debate. The results of some calculations and experiments have indicated that the contribution of twin boundaries to phonon scattering is significant, while other reports have argued that they have a weaker scattering effect than grain boundaries33. It should be noted that most twinned crystals have been reported with coherent twin boundaries, and such coherence is suspected to induce minor thermal boundary resistance. In Zr0.88NiBi, the formation of the secondary nanotwin originates from the defect (kink) on the twin boundary, implying that the compound’s lattice coherence has been interrupted. It is thus reasonable that the twofold twinning configuration also contributes to effective phonon scattering.
More intriguingly, Zr0.88NiBi possesses much lower κlat than Nb0.8CoSb (Fig. 3a) although its vacancy concentration is lower, indicating that there are other underlying mechanisms responsible for its extraordinarily low κlat. The kinetic theory provides a direct correlation between a solid material’s κlat and its phonon relaxation time (determined by scattering) and group velocity34. Since the phonon group velocity in the low-frequency region can be approximated by the sound velocity (vs), the measured vs values of various HHs were compared for further discussion16,35,36,37,38. In a ball-and-spring lattice, vs is governed by the restoring force (F) and the average mass (\(\bar{M}\)) as \({v}_{s} \sim {(\frac{F}{\bar{M}})}^{1/2}\). As shown in Fig. 3b, there is no doubt that heavy-element-containing HHs exhibit lower vs. For instance, the vs values of Zr0.88NiBi and Nb0.8CoSb are 2500 m s−1 and 3313 m s−1, respectively. In addition, the relativistic contraction of the Bi-6s orbital enables inert chemical bonds36 (i.e., a global weak interatomic force). It should also be noted that, regardless of their similar \(\bar{M}\) or the nature of Bi, Zr0.88NiBi is distinguished among the heavy-element-containing HHs by its anomalously low vs. We assumed a much weaker interatomic force in Zr0.88NiBi and then calculated its electron localization function (ELF) and bond lengths to investigate its bonding characteristics (2 × 2 × 2 supercell with removal of one Zr atom corresponding to Zr0.875NiBi, hereafter approximated as Zr0.88NiBi). The ELF value ranges from 0 to 1, where ELF = 1 indicates complete localization of electrons. Figure 3c, d illustrates the projected two-dimensional ELFs along the \((01\bar{1})\) plane for Zr0.88NiBi and ZrCoBi, respectively, in both of which thin gray circle-like contour lines denote regions with the ELF value of 0.8, thus revealing strong electron localization in the Bi atoms for both materials. This phenomenon is similar to that observed in RhBi4, which was attributed to the material’s lone pairs of Bi atoms39. More intriguingly, asymmetric distribution of the electron cloud in the Bi atoms that adjoin Zr vacancies is observed in Zr0.88NiBi, as evidenced by the existence of regions with the higher ELF value of 0.85 (denoted by the thick black crescent-like contour lines in Fig. 3c) in the direction of the vacancies. As a result, a local structural distortion occurs and is accompanied by diverse bond lengths (dNi-Bi = 2.67–2.78 Å and dZr-Ni = 2.68–2.71 Å), in contrast to ZrCoBi, which shows identical Co–Bi and Zr–Co bond lengths (~2.70 Å). Such diversity in bond lengths permits large and anisotropic atomic displacement, which is a typical indicator of soft bonds. Atomic displacement parameters (ADPs) will be discussed later. Both the appearance of asymmetric electron localization in the vacancy-neighboring Bi atoms in Zr0.88NiBi and the slight changes in its Zr–Ni bond lengths indicate the local softening of chemical bonds in this case, and this can also be ascertained from the results of ADP analysis discussed below. Evidently, the nature of Bi and the existence of Zr vacancy are together responsible for the hierarchical chemical bonds (coexistence of global and local soft bonds) in Zr0.88NiBi, which can disrupt phonon propagation and result in much lower vs.

a Comparison of temperature-dependent lattice thermal conductivity among Zr0.88NiBi, ZrCoBi, and Nb0.8CoSb. b Experimentally determined sound velocity of various HHs with respect to their average atomic mass16,35,36,37,38. The gray shaded region shows the trend of \({v}_{s} \sim {(\bar{M})}^{-1/2}\) and the corresponding data of possible HHs are from the AFLOWLIB.org database65 (see details in the Supplemental Material). Calculated two-dimensional ELFs along the \((01\bar{1})\) plane together with corresponding bond lengths for c Zr0.88NiBi and d ZrCoBi. Thin gray circle-like and thick black crescent-like contour lines correspond to the ELF values of 0.8 and 0.85, respectively. Calculated phonon dispersion, PDOS, and ADPs for e Zr0.88NiBi and f ZrCoBi. Ranges of ADPs for all atoms in the supercell of Zr0.88NiBi (see Figure S4) are shown in (e).
To further discern the fantastic phonon dynamics of Zr0.88NiBi, its phonon dispersion, phonon density of states (PDOS), and temperature-dependent ADPs were studied (Fig. 3e). First, no imaginary frequency was observed in the phonon dispersion of Zr0.88NiBi, implying its dynamical stability. In contrast to corresponding results for ZrCoBi (Fig. 3f) and phonon dispersion results for the supercell of Zr8Co8Bi8 (Figure S3), the flattened phonon dispersion and large ADPs of Zr0.88NiBi reinforce our finding of its soft bonding nature since the reduced overall group velocity can be directly visualized by the flattened phonon dispersion and the loosely bound atoms tend to vibrate with larger displacement. The PDOS of Zr0.88NiBi shows that its lattice vibration with a frequency below ~3 THz is mainly dominated by Bi atoms and that the cutoff frequency of its acoustic phonons is ~2.2 THz, which is significantly lower than that for ZrCoBi (approaching 4 THz). Moreover, differing from ZrCoBi with a large acoustic-optical gap, Zr0.88NiBi features low-lying optical phonons (1.5−3 THz) that are strongly hybridized with acoustic branches (in the frequency range between 1.87 and 2.22 THz). We expect that these low-lying optical phonons originate from the compound’s local soft bonds, and an analogous phenomenon was also reported for α-MgAgSb40. Importantly, these soft optical modes with nearly vanishing group velocities show a non-propagating-like behavior while enabling additional scattering for heat-carrying acoustic phonons. The prevailing viewpoint is that thermal resistance results from the lowest-order three-phonon scattering, which gives rise to a T−1 dependence of κlat34. However, a higher-order phonon scattering process considering optical phonons has gained increasing attention and its adequacy has also been proved in other materials such as boron arsenide41,42,43 and hybrid perovskites44,45.
It should be noted that there are visible fluctuations in the ADPs for each atom in the supercell of Zr0.88NiBi, as shown in Fig. 3e with details along the three Cartesian directions plotted in Figure S4, which is attributed to the diversity of bond lengths. The ratios of the largest to smallest ADPs for Zr, Ni, and Bi are 1.24:1, 1.12:1, and 1.65:1, respectively, at room temperature. Moreover, the smallest ADP for Bi in Zr0.88NiBi shows only a minor difference compared to the ADP for Bi in ZrCoBi, indicating the similar vibrating modes in these two materials governed by a global soft bond. The fact that the largest and second-largest ADPs respectively correspond to Bi5 and Bi4 (both along the y-axis), which both show an asymmetrically distributed electron cloud adjacent to a Zr vacancy (see Fig. 3c), attests to the existence of a vacancy-induced much softer local chemical bond. In addition, decent lattice anharmonicity in Zr0.88NiBi is evidenced by its anisotropic ADPs, especially at ambient temperature. Such anisotropic behavior is usually absent in highly symmetrical HHs (e.g., identical ADPs along the three Cartesian directions for ZrCoBi) and is also correlated to the local soft bonds in the case of Zr0.88NiBi. Therefore, our in-depth analysis of vacancy-mediated anomalous phononic transport in Zr0.88NiBi successfully elucidates the origin of its intrinsically low κlat, which is attributed to the multiple phonon scattering processes in the compound and its global and local soft bonding environment.
Electronic properties
Given the multifunctionality of vacancy in tailoring phononic properties, as well as the potential for high thermoelectric performance, the electronic properties of Zr0.88NiBi deserve further attention. The electron-sufficient configuration of Zr0.88NiBi results in n-type self-doping behavior, which can also be identified by the negative Seebeck coefficient (S). As seen in the unfolded electronic band structure of Zr0.88NiBi (Fig. 4a), the Fermi level lies well above the conductive band minimum (CBM) with an electron concentration of ~9.89 × 1021 cm−3 and the CBM locates at the X point with three-fold valley degeneracy, which is analogous to most 18-electron HHs. By plotting the Pisarenko relation as shown in Fig. 4b, the modeled density of states (DOS) effective mass is ~6.0 me (details for the single parabolic band model are provided in the Supplemental Material). It should be noted that the CBM of a HH is primarily governed by the interaction between the d-orbitals of two transition metals46 (e.g., Zr and Ni in the compound studied here). This orbital configuration should give rise to a similar conductive band structure among different compounds when the two transition metals are unchanged, such as the comparable DOS effective mass values of ZrNiSn and ZrNiPb (~3.0 me)47,48. In addition, ZrCoSb and ZrCoBi also display identical behavior49,50, further confirming the negligible influence of the main group element on the CBM. The larger \({m}^{*}\) of Zr0.88NiBi thus seems counterintuitive, but the flat band near the CBM along the Γ and X directions that exists in Zr0.88NiBi is not observed in either ZrNiSn or ZrNiPb. This flat band probably arises from the localized orbital around the vacancy, resembling HfO2, in which the vacancy causes a local spin state and unexpected ferromagnetism6. Moreover, based on the theory of topological quantum chemistry51, the nature of symmetry-enforced Fermi degeneracy at high-symmetry points qualifies this material as a type of topological electronic material, analogous to YPdBi documented in ref. 52. We thus speculate that this material could attract considerable attention for condensed matter physics research due to its exotic band structure. We further estimated the mean free path of an electron at ~6.33 Å based on the formula \({mfp}=\sqrt{2{E}_{F}{m}_{b}^{*}}\frac{\mu }{e}\), where EF, \({m}_{b}^{*}\), μ, and e are the Fermi energy, effective band mass (\({m}^{*}={N}_{V}^{2/3}{m}_{b}^{*}\)), mobility, and elementary electron charge, respectively. The mfp of Zr0.88NiBi is comparable to its lattice constant (~6.19 Å), suggesting that its electron transport has reached the Ioffe–Regel limit53, which is reasonable given its unconventionally defective structure. This also implies that the carrier filtering effect from twin boundaries could be negligible since electrons are scattered before this effect occurs.

a Calculated unfolded electronic band structure of Zr0.88NiBi. b Pisarenko relation of Zr0.88NiBi in comparison to that of ZrNiSn47, ZrNiPb48, ZrCoSb49, and ZrCoBi50. Symbols: experimental data. Dashed lines: obtained from the SPB model. Temperature-dependent c Seebeck coefficient and d electrical conductivity of Zr0.88Ni1-xCoxBi. e Comparison of the electronic contribution to total heat conduction between Zr0.88NiBi (top panel) and Zr0.88Ni0.57Co0.43Bi (bottom panel). f Composition-dependent electronic thermal conductivity and power factor (top panel) and reduced Fermi energy (bottom panel) for Zr0.88Ni1-xCoxBi.
Nonetheless, a deep Fermi level location is unfavorable to good thermoelectric performance. Hong et al. specified that the optimal position of the Fermi level is near the band edge54. To downshift the Fermi level in the Zr0.88NiBi-based compound, acceptor cobalt was thus doped for electron capture. The temperature-dependent S of Zr0.88Ni1-xCoxBi is presented in Fig. 4c. In accordance with the inverse correlation between S and carrier concentration, |S| increases with increasing doping concentration (x), suggesting that cobalt provides efficient electron capture. As a result, the electron concentration is reduced to ~1.48 × 1021 cm−3 for Zr0.88Ni0.57Co0.43Bi. Electrical conductivity (σ) decreases with increasing x, also in accordance with the reduced electron concentration (Fig. 4d). Since electronic thermal conductivity (κele) is determined by the carrier concentration as well, high κele is a major drawback of Zr0.88NiBi that can be addressed via the efficient electron capture provided by cobalt doping. As shown in the top panel of Fig. 4e, the contribution to total thermal conductivity (κtot) from electrons is ~63% at room temperature and, following the Wiedemann-Franz law, becomes more significant with increasing temperature. On the other hand, the electronic contribution is significantly suppressed in Zr0.88Ni0.57Co0.43Bi (bottom panel, Fig. 4e), such that phonons have the dominant role in heat conduction (the phononic contribution can be found in Figure S5). Consequently, cobalt doping enables synergistic optimization of both κele and power factor (PF, the product S2σ), which is a unique advantage over the typical 18-electron HHs. Most 18-electron HHs are intrinsic semiconductors and optimization of their performance thus relies on an elevated carrier concentration that will inevitably lead to a monotonically increased κele. The countertrend behavior of the compound studied here is illustrated in the top panel of Fig. 4f. We further determined the composition-dependent reduced Fermi energy (\({\xi }_{F}=\frac{{E}_{F}}{{k}_{B}T}\), where kB is the Boltzmann constant), and the results are shown in the bottom panel of Fig. 4f. The band-edge-approaching ξF observed for Zr0.88Ni0.57Co0.43Bi is in line with expectations given the corresponding improvement in thermoelectric performance.
Figure of merit
Benefitting from the efficient electron capture discussed above, the peak ZT of Zr0.88Ni0.57Co0.43Bi is five times that of the undoped Zr0.88NiBi (Fig. 5a). By further alloying isovalent antimony at the bismuth site, a peak ZT of ~1.0 at 973 K is obtained for Zr0.88Ni0.57Co0.43Bi0.9Sb0.1 (the thermoelectric properties of Zr0.88Ni0.57Co0.43Bi1-ySby are shown in Figure S6; additionally, the phase purity of all Zr0.88Ni1-xCoxBi1-ySby samples studied is shown in Figure S1c). The performance advantage of this material over other typical n-type HHs is evidenced by the comparisons shown in Fig. 5b, c16,18,35,38,49,50,55,56,57,58. Although the archetypal ZrNiSn-based HH exhibits similar ZT values at lower temperatures, the bipolar transport effect causes its performance to degrade at high temperature, making it incompatible with p-type HHs working at 973 K. In comparison to the recently reported n-type ZrCoBi-based HH with high performance at 973 K, an overall enhanced ZT can be observed in the material studied here. Consequently, Zr0.88Ni0.57Co0.43Bi0.9Sb0.1 exhibits a decent average ZT (over the range of 300–973 K), outperforming various defective 19-electron and typical 18-electron HHs.

Discussion
Given that defects have been regarded as an additional degree of freedom for boosting thermoelectric performance, studies of the vacancy-abundant half-Heuslers have the potential to reshape the development of high-temperature thermoelectric materials. Here we modeled the figure of merit of the defective ZrNiBi at 973 K. When the electron transport of defective ZrNiBi reaches the Ioffe–Regel limit (electron mean free path ~a, where a denotes the lattice constant)53 and its phonon transport follows the Cahill model (minimal lattice thermal conductivity ~0.66 W m−1 K−1)59, its figure of merit exhibits an impressive value of ~2.2 at the electron concentration of 4–5 × 1020 cm−3 (Figure S7a). It should be noted that our transmission electron microscope results revealed a random distribution of vacancies in Zr0.88NiBi, in contrast to some typical defective 19-electron half-Heuslers that have been reported to show short-range order among their vacancies (e.g., Nb0.8CoSb22). It is thus natural to examine whether the performance of defective ZrNiBi could be enhanced if its degree of vacancy order can be improved. However, the existence of vacancy pairs and vacancy triplets complicates the short-range order in defective 19-electron half-Heusler systems22. Since long-range order is an extreme case of vacancy order, we assumed the existence of long-range-ordered vacancies in Zr0.88NiBi (where the shortest inter-vacancy distance of \(\sqrt{2}a\) is assigned to the electron mean free path) and, correspondingly, the material’s figure of merit as a function of both lattice thermal conductivity and electron concentration was plotted in Figure S7b. In comparison to the framework of the Ioffe–Regel condition, the optimal electron concentration is shifted to a lower range and the material exhibits better performance, e.g., its peak figure of merit is enhanced to ~2.7 as the lattice thermal conductivity decreases to the value estimated by the Cahill model. It must be noted that a system with only long-range-ordered vacancies is hardly achievable based on experimental results, so the trend found from our model sets an upper bound for the thermoelectric performance of defective ZrNiBi while the degree of vacancy order can serve as a control for its figure of merit. In fact, current optimization approaches have resulted in a peak figure of merit of ~1.0 for a Zr0.88NiBi-based material at a lattice thermal conductivity of ~0.9 W m−1 K−1 and an electron concentration of ~1.5 × 1021 cm−3. To fulfill its thermoelectric potential, delicate phonon and electron concentration engineering (toward lower values for both lattice thermal conductivity and electron concentration) is necessary. In addition, vacancy manipulation for ordered distribution enables further enhancement of performance as depicted in Figure S7c.
In summary, an emerging defective ZrNiBi-based compound with distinctive structural characteristics and anomalous transport properties that distinguish it among the half-Heuslers is introduced. Its randomly distributed cationic vacancies and twofold twins were directly visualized using real-space imaging. Phonon dynamics analysis demonstrated that vacancy-induced point defect scattering, low-lying optical phonons, anisotropic vibrations, and local soft bonds, going beyond the conventional vacancy scattering model, together correlate highly with the compound’s extraordinarily low lattice thermal conductivity (~1.4 W m−1 K−1 at room temperature). Regarding its electronic properties, experimental Pisarenko relations and first-principles calculations confirmed the existence of a flat band near the conductive band minimum that is also vacancy related and has been rarely reported in other half-Heuslers. Together with the symmetry-enforced Fermi degeneracy at high-symmetry points, the exotic band structure makes defective ZrNiBi quite appealing for condensed matter physics research. With the benefits of vacancy-mediated anomalous transport properties and optimized downshifting of the Fermi level to the band edge, significantly enhanced thermoelectric performance was achieved in a defective ZrNiBi-based material, outperforming various typical n-type half-Heuslers. We expect that the discovery of this promising thermoelectric material could further advance the potential of thermoelectric half-Heuslers and that our findings of vacancy-mediated anomalous transport properties will trigger in-depth research on other fascinating defect-driven phenomena.
Methods
Material synthesis
The phase-pure ZrNiBi-based compounds were synthesized by ball-milling, hot-pressing, and annealing processes with high-purity raw elements (Zr sponges, 99.2%; Ni powder, 99.8%; Bi pieces, 99.99%; Co powder, 99.8%; and Sb shots, 99.8%). The raw elements were stoichiometrically weighed and then loaded into an argon-filled stainless steel jar for the subsequent ball-milling process. After 20 h of ball-milling, each obtained nanoscale powder sample was hot-pressed at ~1000 K with a holding time of 2 min. Each resulting solidified disk was sealed in a quartz tube and then annealed at 973 K for 40 h. The heating rates for hot-pressing and annealing were ~150 K/min and 3.8 K/min, respectively.
Structure characterization
X-ray diffraction (XRD, Rigaku SmartLab diffractometer) was performed to identify the phase composition of each sample. Transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) investigations were carried out using a double-Cs corrected JEM-ARM200F equipped with a cold field emission gun (FEG) source. Specimens used for TEM observation were prepared by traditional mechanical polishing, dimpling, and subsequent ion milling with a liquid nitrogen stage. All the HAADF images in this study were Fourier-filtered to reduce the influence of random noise.
Transport measurement
The Seebeck coefficient (S) and electrical conductivity (σ) of each sample were simultaneously measured using a commercial system (ZEM-3, ULVAC). Hall carrier concentration (nH) was obtained using a physical properties measurement system (PPMS, Quantum Design) under a magnetic field of ±7 T and electrical current of 8 mA. Thermal conductivity (κtot) was calculated based on κtot = DCPρ, where D is thermal diffusivity measured by laser flash (LFA 457, Netzsch), CP is the specific heat obtained by the Dulong-Petit law, and ρ is mass density determined by an Archimedes’ method. Electronic thermal conductivity (κele) was calculated based on κele = LσT, where L is the Lorenz number deduced by the single parabolic band model and T is the absolute temperature. Lattice thermal conductivity (κlat) was determined by subtracting the κele from κtot. Sound velocity (vs) studies were carried out using a RITEC RAM-5000 Advanced Ultrasonic Measurement System, which incorporates a pulse-echo technique for time propagation measurements. Piezoelectric transducers (Olympus NDT, Inc.) operating at a frequency of 10 MHz were used to generate and receive longitudinal and shear ultrasonic bulk waves. Propylene glycol and shear wave couplant (SWC) (both from Olympus NDT Inc.) were used as couplant materials for longitudinal and shear modes, respectively. The time resolution achieved in our experiments was better than 0.2 ns.
Theoretical calculation
First-principles density functional theory (DFT) calculations were carried out using the Vienna ab initio Simulation Package (VASP) with the projector augmented wave (PAW) scheme60. The Perdew, Burke, and Ernzerhof (PBE) electronic exchange correlation functional was used under a gradient-generalized approximation (GGA)61. We used a constant energy cutoff of 500 eV to truncate the plane-wave basis and a roughly constant density of k-points (30 Å3) of Monkhorst-Pack62 point meshes to sample the Brillouin zones. All structures were relaxed with respect to all the forces, and components of the tensors were below 0.01 eV/Å and 0.2 kbar, respectively. A 2 × 2 × 2 supercell with 23 atoms following removal of one Zr atom (i.e., Zr7Ni8Bi8) was used to calculate the electronic structure of Zr0.875NiBi. To determine the band structure, the band unfolding method (BandUP code)63 was used to unfold the band structure of the supercell into its primitive Brillouin zone. Phonon dispersions of Zr0.875NiBi and ZrCoBi were calculated using 2 × 2 × 2 supercells with 184 atoms and 3 × 3 × 3 supercells with 81 atoms, respectively, by the finite displacement method implemented in the Phonopy package64.
Data availability
All data are available in the main text and the Supplementary Materials.
References
Lu, Q. et al. Bi-directional tuning of thermal transport in SrCoOx with electrochemically induced phase transitions. Nat. Mater. 19, 655–662 (2020).
Chen, C. et al. Atomic line defects and zero-energy end states in monolayer Fe(Te,Se) high-temperature superconductors. Nat. Phys. 16, 536–540 (2020).
Hanson, R. Diamond defects cooperate via light. Science 354, 835–836 (2016).
Avsar, A. et al. Defect induced, layer-modulated magnetism in ultrathin metallic PtSe2. Nat. Nanotechnol. 14, 674–678 (2019).
Venkatesan, M., Fitzgerald, C. B. & Coey, J. M. Unexpected magnetism in a dielectric oxide. Nature 430, 630 (2004).
Bouzerar, G. & Ziman, T. Model for vacancy-induced d0 ferromagnetism in oxide compounds. Phys. Rev. Lett. 96, 207602 (2006).
Dutt, M. V. et al. Quantum register based on individual electronic and nuclear spin qubits in diamond. Science 316, 1312–1316 (2007).
Lozovoi, A. et al. Optical activation and detection of charge transport between individual colour centres in diamond. Nat. Electron. 4, 717–724 (2021).
Jo, J. et al. Defect-gradient-induced Rashba effect in van der Waals PtSe2 layers. Nat. Commun. 13, 2759 (2022).
Tan, G. et al. High thermoelectric performance SnTe-In2Te3 solid solutions enabled by resonant levels and strong vacancy phonon scattering. Chem. Mater. 27, 7801–7811 (2015).
Mao, J. et al. Self-compensation induced vacancies for significant phonon scattering in InSb. Nano Energy 48, 189–196 (2018).
Yao, H. et al. Vacancy ordering induced topological electronic transition in bulk Eu2ZnSb2. Sci. Adv. 7, eabd6162 (2021).
Wu, Y. et al. Lattice strain advances thermoelectrics. Joule 3, 1276–1288 (2019).
Zeier, W. G. et al. Using the 18-electron rule to understand the nominal 19-electron half-Heusler NbCoSb with Nb vacancies. Chem. Mater. 29, 1210–1217 (2017).
Anand, S. et al. A valence balanced rule for discovery of 18-electron half-Heuslers with defects. Energy Environ. Sci. 11, 1480–1488 (2018).
Xia, K. et al. Enhanced thermoelectric performance in 18-electron Nb0.8CoSb half-Heusler compound with intrinsic Nb vacancies. Adv. Funct. Mater. 28, 1705845 (2018).
Anand, S., Xia, K., Zhu, T., Wolverton, C. & Snyder, G. J. Temperature dependent n‐type self doping in nominally 19‐electron half‐Heusler thermoelectric materials. Adv. Energy Mater. 8, 1801409 (2018).
Fang, T. et al. A new defective 19-electron TiPtSb half-Heusler thermoelectric compound with heavy band and low lattice thermal conductivity. Mater. Today Phys. 13, 100200 (2020).
Graf, T., Felser, C. & Parkin, S. S. P. Simple rules for the understanding of Heusler compounds. Prog. Solid State Chem. 39, 1–50 (2011).
Gautier, R. et al. Prediction and accelerated laboratory discovery of previously unknown 18-electron ABX compounds. Nat. Chem. 7, 308–316 (2015).
Zunger, A. Inverse design in search of materials with target functionalities. Nat. Rev. Chem. 2, 1–16 (2018).
Xia, K. et al. Short-range order in defective half-Heusler thermoelectric crystals. Energy Environ. Sci. 12, 1568–1574 (2019).
Ren, W. Y., Shi, X., Wang, Z. M. & Ren, Z. F. Crystallographic design for half-Heuslers with low lattice thermal conductivity. Mater. Today Phys. 25, 100704 (2022).
Kirklin, S. et al. The Open Quantum Materials Database (OQMD): assessing the accuracy of DFT formation energies. NPJ Comput. Mater. 1, 15010 (2015).
Zhu, Q. et al. Hierarchical twinning governed by defective twin boundary in metallic materials. Sci. Adv. 8, eabn8299 (2022).
Mao, J., Wang, Y. M., Liu, Z. H., Ge, B. & Ren, Z. F. Phonon scattering by nanoscale twin boundaries. Nano Energy 32, 174–179 (2017).
Liu, Z. H. et al. The influence of doping sites on achieving higher thermoelectric performance for nanostructured α-MgAgSb. Nano Energy 31, 194–200 (2017).
Smith, T. M. et al. Phase transformation strengthening of high-temperature superalloys. Nat. Commun. 7, 13434 (2016).
Wu, Q. et al. Phase-selective recrystallization makes eutectic high-entropy alloys ultra-ductile. Nat. Commun. 13, 4697 (2022).
Lu, K. Stabilizing nanostructures in metals using grain and twin boundary architectures. Nat. Rev. Mater. 1, 16019 (2016).
Zhang, R. et al. Short-range order and its impact on the CrCoNi medium-entropy alloy. Nature 581, 283–287 (2020).
Klemens, P. G. Thermal conductivity and lattice vibrational modes. Solid State Phys. 7, 1–98 (1958).
Mao, J. et al. Advances in thermoelectrics. Adv. Phys. 67, 69–147 (2018).
Qian, X., Zhou, J. & Chen, G. Phonon-engineered extreme thermal conductivity materials. Nat. Mater. 20, 1188–1202 (2021).
He, R. et al. Enhanced thermoelectric properties of n-type NbCoSn half-Heusler by improving phase purity. APL Mater. 4, 104804 (2016).
Zhu, H. T. et al. Discovery of ZrCoBi based half Heuslers with high thermoelectric conversion efficiency. Nat. Commun. 9, 2497 (2018).
Zhu, H. T. et al. Discovery of TaFeSb-based half-Heuslers with high thermoelectric performance. Nat. Commun. 10, 270 (2019).
Li, S. et al. Titanium doping to enhance thermoelectric performance of 19‐electron VCoSb half‐Heusler compounds with vanadium vacancies. Ann. Phys. 532, 1900440 (2020).
Grin, Y., Wedig, U. & von Schnering, H. G. Hyperbolic lone pair structure in RhBi4. Angew. Chem. Int. Ed. 34, 1204–1206 (1995).
Ying, P. et al. Hierarchical chemical bonds contributing to the intrinsically low thermal conductivity in α-MgAgSb thermoelectric materials. Adv. Funct. Mater. 27, 1604145 (2017).
Feng, T., Lindsay, L. & Ruan, X. Four-phonon scattering significantly reduces intrinsic thermal conductivity of solids. Phys. Rev. B 96, 161201 (2017).
Tian, F. et al. Unusual high thermal conductivity in boron arsenide bulk crystals. Science 361, 582–585 (2018).
Li, S. et al. Anomalous thermal transport under high pressure in boron arsenide. Nature 612, 459–464 (2022).
Ma, H. et al. Supercompliant and soft (CH3NH3)3Bi2I9 crystal with ultralow thermal conductivity. Phys. Rev. Lett. 123, 155901 (2019).
Li, C. et al. Remarkably weak anisotropy in thermal conductivity of two-dimensional hybrid perovskite butylammonium lead iodide crystals. Nano Lett. 21, 3708–3714 (2021).
Zeier, W. G. et al. Engineering half-Heusler thermoelectric materials using Zintl chemistry. Nat. Rev. Mater. 1, 16032 (2016).
Xie, H. H. et al. The intrinsic disorder related alloy scattering in ZrNiSn half-Heusler thermoelectric materials. Sci. Rep. 4, 6888 (2014).
Mao, J. et al. Thermoelectric properties of n-type ZrNiPb-based half-Heuslers. Chem. Mater. 29, 867–872 (2017).
Ren, W. Y. et al. Intermediate-level doping strategy to simultaneously optimize power factor and phonon thermal conductivity for improving thermoelectric figure of merit. Mater. Today Phys. 15, 100250 (2020).
Zhu, H. T. et al. Understanding the asymmetrical thermoelectric performance for discovering promising thermoelectric materials. Sci. Adv. 5, eaav5813 (2019).
Bradlyn, B. et al. Topological quantum chemistry. Nature 547, 298–305 (2017).
Vergniory, M. G. et al. All topological bands of all nonmagnetic stoichiometric materials. Science 376, eabg9094 (2022).
Gurvitch, M. Ioffe-Regel criterion and resistivity of metals. Phys. Rev. B 24, 7404–7407 (1981).
Hong, M., Lyu, W., Wang, Y., Zou, J. & Chen, Z. G. Establishing the golden range of seebeck coefficient for maximizing thermoelectric performance. J. Am. Chem. Soc. 142, 2672–2681 (2020).
Huang, L. et al. Enhanced thermoelectric performance of nominal 19-electron half-Heusler compound NbCoSb with intrinsic Nb and Sb vacancies. Mater. Today Phys. 20, 100450 (2021).
Zhang, H. et al. Thermoelectric properties of n-type half-Heusler compounds (Hf0.25Zr0.75)1-xNbxNiSn. Acta Mater. 113, 41–47 (2016).
Li, S. et al. N-type TaCoSn-based half-Heuslers as promising thermoelectric materials. ACS Appl. Mater. Interfaces 11, 41321–41329 (2019).
Liu, Y. et al. Lanthanide contraction as a design factor for high-performance half-Heusler thermoelectric materials. Adv. Mater. 30, 1800881 (2018).
Cahill, D. G., Watson, S. K. & Pohl, R. O. Lower limit to the thermal conductivity of disordered crystals. Phys. Rev. B 46, 6131–6140 (1992).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Medeiros, P. V. C., Stafström, S. & Björk, J. Effects of extrinsic and intrinsic perturbations on the electronic structure of graphene: retaining an effective primitive cell band structure by band unfolding. Phys. Rev. B 89, 041407 (2014).
Gonze, X. & Lee, C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 55, 10355–10368 (1997).
Curtarolo, S. et al. AFLOW: an automatic framework for high-throughput materials discovery. Comput. Mater. Sci. 58, 218–226 (2012).
Acknowledgements
Z.M.W. acknowledges financial support from the National Key Research and Development Program of China (2019YFB2203400) and the “111 Project” (B20030). W.Y.R. acknowledges financial support from the Sichuan Science and Technology Program (2023NSFSC0970). J.W. acknowledges financial support from the Innovation Group Project of Sichuan Province (20CXTD0090). R.H. acknowledges financial support from the Deutsche Forschungsgemeinschaft (DFG, 453261231). J.v.d.B. acknowledges the Deutsche Forschungsgemeinschaft (DFG) for support through the Wrüzburg-Dresden Cluster of Excellence on Complexity and Topology in Quantum Matter ct.qmat (EXC 2147, Project No. 39085490) and the Collaborative Research Center SFB 1143 (Project No. 247310070). C.W.C. acknowledges financial support from the U.S. Air Force Office of Scientific Research Grants FA9550-15-1-0236 and FA9550-20-1-0068, the T.L.L. Temple Foundation, and the John J. and Rebecca Moores Endowment. S.G. thanks Oleg Janson for fruitful discussions and Ulrike Nitzsche at IFW Dresden for technical assistance. We also thank Troy Christensen at the University of Houston for revising the paper and Professor David J. Singh at University of Missouri for discussions.
Author information
Authors and Affiliations
Contributions
W.Y.R., Z.M.W., and Z.F.R. designed this work. W.Y.R. and S.W.S. synthesized the samples. W.Y.R., R.H., L.Z.D., and A.S. conducted the transport property measurements. W.Y.R., W.H.X., and Y.M.W. performed the structure characterizations. S.P.G. and J.v.d.B. did the theoretical calculations. K.N., J.W., and C.W.C. helped with the transport property analysis. G.H.G., S.C., and Y.M.H. helped with the TEM result analysis. W.Y.R. and Z.F.R. wrote the manuscript, and all authors edited it.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xun Shi, Yuan Yu and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ren, W., Xue, W., Guo, S. et al. Vacancy-mediated anomalous phononic and electronic transport in defective half-Heusler ZrNiBi. Nat Commun 14, 4722 (2023). https://doi.org/10.1038/s41467-023-40492-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-40492-7
This article is cited by
-
Vacancies tailoring lattice anharmonicity of Zintl-type thermoelectrics
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
