
Abstract
The ability to dynamically manipulate the transcriptome is important for studying how gene networks direct cellular functions and how network perturbations cause disease. Nuclease-dead CRISPR–dCas9 transcriptional regulators, while offering an approach for controlling individual gene expression, remain incapable of dynamically coordinating complex transcriptional events. Here, we describe a flexible dCas9-based platform for chemical-inducible complex gene regulation. From a screen of chemical- and light-inducible dimerization systems, we identified two potent chemical inducers that mediate efficient gene activation and repression in mammalian cells. We combined these inducers with orthogonal dCas9 regulators to independently control expression of different genes within the same cell. Using this platform, we further devised AND, OR, NAND, and NOR dCas9 logic operators and a diametric regulator that activates gene expression with one inducer and represses with another. This work provides a robust CRISPR–dCas9-based platform for enacting complex transcription programs that is suitable for large-scale transcriptome engineering.
Similar content being viewed by others

Main
The cellular transcriptome is a convergence point that integrates myriad signals to execute biological processes. During development, the transcriptional programs of stem cells coordinate dynamic changes to the mRNA abundance of key genes, driving differentiation into various cell types. Transcriptome disruption is a hallmark of disease. Genome-wide association studies demonstrate that the majority (>93%) of cellular trait- and disease-associated variants exist within noncoding DNA, highlighting the essential role of transcriptional regulation in these processes1,2. While techniques such as DNA microarrays and RNA-seq enable reading of the cellular transcriptome3,4, researchers must be able to manipulate transcription in a multiplexed and dynamic fashion in order to establish causal relationships between gene networks and cellular function. Conventional small-molecule-inducible transgene expression systems based on doxycycline or ponasterone A allow manipulation of individual genes5,6, yet they lack the flexibility and scalability required for simultaneous multigene modulation. Repurposing genome engineering technologies may offer a promising solution for flexible transcriptome engineering of complex gene networks.
The bacterial CRISPR–Cas9 system is a versatile RNA-guided platform for genome engineering in cell culture or in vivo systems7. Nuclease Cas9 protein, when paired with a single guide RNA (sgRNA), can be used to precisely edit genomic DNA8,9,10,11. Beyond editing, nuclease-dead Streptococcus pyogenes Cas9 (D10A and H840A), termed dCas9, can be combined with transcriptional activation or repression effectors for sequence-specific constitutive activation or gene repression in mammalian systems12,13,14,15,16,17. Furthermore, proof-of-concept work has shown that dCas9 can be fused to light- and chemical-inducible domains to conditionally activate gene expression using extrinsic signals18,19,20. While these inducible dCas9 transcriptional regulators enable regulation of individual genes in isolation, engineering dCas9 regulators that simultaneously utilize multiple inducible systems remains an unexplored area of research. If such inducible systems can act without crosstalk in the same cell, then they can be used in parallel on different dCas9s to dynamically regulate independent gene targets or in combination on a single dCas9 to regulate one target using multiple signals. Given the flexibility and ease of use of CRISPR–Cas9, such advances could provide an approach for the complex regulation of gene expression and for the study of sophisticated gene networks. In addition, the resulting regulators would expand the toolkit of functional parts available for building dCas9-based synthetic gene circuits in mammalian cells21.
Here, we present a dCas9-based platform for engineering inducible and orthogonal transcriptional regulators to produce complex transcriptional programs. We systematically explore how dCas9 can be coupled with chemical- and light-inducible domains to dynamically regulate gene activation and repression. These technologies enable the manipulation of multiple gene targets in distinct regulatory patterns and can be utilized for applications requiring reversible and highly specific control over gene transcription such as developmental and disease modeling.
Results
Screening chemical- or light-inducible dCas9s for efficient gene activation
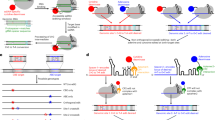
We designed an inducible dCas9-based transcriptional regulation system in which dCas9 and a transcriptional effector are fused to complementary pairs of heterodimerization domains and only assemble in the presence of an extrinsic inducer. To test this design, we systematically fused S. pyogenes (Sp) dCas9 and a VPR (VP64-p65-Rta) activator22 to six previously reported chemical- and light-inducible heterodimerization domains cloned onto a dual-promoter PiggyBac vector (Supplementary Fig. 1a). These heterodimerization domains consisted of abscisic acid (ABA)-inducible ABI–PYL1 (ref. 23), gibberellin (GA)-inducible GID1–GAI24, rapamycin-inducible FKBP–FRB25, phytochrome-based red light-inducible PHYB–PIF26, cryptochrome-based blue light-inducible CRY2PHR–CIBN27, and light oxygen voltage-based blue-light-inducible FKF1–GI28.
We assessed the activation efficiency of each inducible dCas9 in HEK293T cells by targeting a Tet-On promoter (pTRE3G)–EGFP reporter (Supplementary Fig. 1b). The most potent systems were the ABA and GA heterodimerization systems, both derived from plant hormone signaling pathways23,24. Upon induction, the ABA- and GA-dimerized VPR–Sp-dCas9-activated EGFP protein expression 165- and 94-fold, respectively (Fig. 1a,b), efficiencies comparable to that of a VPR–Sp dCas9 direct fusion protein (Supplementary Fig. 2a). Critically, these inducible dCas9s showed no leaky activation when expressed without inducer. Rather, we observed a minor decrease (10–30%) in EGFP relative to reporter-only cells (Fig. 1a,b), an effect likely due to steric interference with native transcription factors from dCas9 occupancy on the promoter12. Furthermore, addition of inducers to reporter cells without dCas9 elicited no increase in EGFP relative to uninduced cells, excluding nonspecific activation. In contrast, the three light-inducible systems and the rapamycin system showed modest EGFP activation (1.3- to 5.6-fold) upon induction (Supplementary Fig. 1c–f). Previous studies found that light-inducible dimerizing domains exhibit low light sensitivity29 and highly context-dependent structural folding30, which may explain the weak response. In addition, we tested exchanging dimerization domains between the dCas9 and effector domains and found that many inverted dCas9 activators showed poor activity (Supplementary Fig. 3). These results suggest that dimerization of the inducible domains may be sensitive to their spatial orientation relative to dCas9.

(a,b) Fluorescence quantification after 48 h induction for HEK293T pTRE3G–EGFP cells transfected with Sp sgTRE3G and (a) ABA-inducible or (b) GA-inducible VPR–Sp dCas9. (c,d) Fluorescence quantification after 5 d induction for HEK293T pSV40–EGFP cells transfected with Sp sgSV40 and (c) ABA-inducible or (d) GA-inducible Sp KRAB–dCas9. Inverse fold-change repression is indicated in magenta. (e,f) Fluorescence quantification after 48 h for HEK293T pTRE3G–EGFP cells transfected with Sa sgTRE3G and (e) ABA-inducible or (f) GA-inducible VPR-Sa dCas9. (g) Immunofluorescence quantification of CXCR4 after 48 h induction in HEK293T cells transfected with Sp sgCXCR4 and ABA-inducible dCas9 fused to a VPR, SunTag–VP64, or SunTag–VPR activation domain. For the no-sgRNA controls in (g), the data represent two independent transfections performed in technical replicates (n = 4); for other conditions in (g) and all other experiments, the data represent four independent transfections performed in technical replicates (n = 8). Mean fluorescence intensities are presented as arbitrary units (a.u.). P values from Games–Howell post hoc tests following Welch's ANOVA are provided in Supplementary Table 1.
Implementing inducible repression of gene expression
To expand the inducible system to gene repression, we replaced the effector fused to the ABA- or GA-inducible dimerization domains with the Krüppel-associated box (KRAB) repression domain31. We assessed inducible repression in HEK293T cells by targeting a constitutive SV40 promoter (pSV40)–EGFP reporter (Supplementary Fig. 4a). The ABA- and GA-inducible systems repressed EGFP 5.6- and 3.2-fold, respectively (Fig. 1c,d), with efficiencies comparable to that of a KRAB–Sp dCas9 direct fusion protein (Supplementary Fig. 2b). Binding of dCas9 alone to pSV40 in uninduced cells showed no or only slight reduction of EGFP signals relative to reporter-only cells for either inducible system (Fig. 1c,d). These results suggest that promoter targeting provides a viable strategy for inducible repression by maximizing the induced repressive effect from KRAB recruitment while minimizing the basal repressive effect of steric hindrance from promoter-bound dCas9 alone.
Staphylococcus aureus inducible dCas9 transcriptional regulators
Recent work showed that a Cas9 protein derived from S. aureus is capable of gene editing32 and transcriptional activation33,34 in mammalian cells. We tested whether our inducible fusion protein design would transfer to nuclease-dead S. aureus Cas9 (Sa dCas9). Both ABA- and GA-inducible activators using Sa dCas9 increased EGFP expression in HEK293T pTRE3G–EGFP cells (Fig. 1e,f) and achieved similar levels of activation as direct fusion VPR–Sa dCas9 (Supplementary Fig. 2c). However, Sa dCas9 activated EGFP to 20–30% the levels of Sp dCas9 overall, possibly because of differences in sgRNA targeting sequence efficiencies, the lack of a functionally optimized Sa sgRNA scaffold35, or intrinsic differences between dCas9 DNA-binding affinities. Nonetheless, retention of gene activation indicates that our fusion design can transfer to multiple Cas9 orthologs.
Inducible activation of endogenous genes
We further validated the activity of ABA-inducible Sp dCas9 on an endogenous gene. Here, we introduced individual dCas9 constructs into HEK293T cells along with an sgRNA targeting the CXCR4 gene promoter. We compared activation using the VPR, SunTag–VP6436, or combined SunTag–VPR effectors (Fig. 1g). For these effectors, ABA induction increased CXCR4 protein levels 6.4- to 8.9-fold relative to the levels of uninduced cells. The inability of the combined SunTag–VPR effector to further increase activation efficiency is corroborated by a recent comparative study of multiple Cas9 activator systems37.
Reversibility and dose responsiveness of ABA- and GA-inducible regulation
We subsequently performed detailed dynamic characterizations of the ABA and GA systems. For activation, we monitored EGFP levels over 7 d in HEK293T pTRE3G–EGFP cells stably expressing ABA- or GA-inducible Sp VPR–dCas9 as well as in cells expressing the Sa counterparts. In all systems tested, cells under continuous inducer treatment over 7 d maintained high levels of EGFP (Fig. 2a,b). Removal of the inducer after 2 d triggered a progressive decrease in EGFP levels to baseline, demonstrating reversibility of activation. Furthermore, readdition of inducer in these cells activated EGFP as strongly and rapidly as the original treatment. GA-inducible systems exhibited slower reversal kinetics upon inducer removal (Fig. 2b), possibly on account of residual GA in the media or slow dissociation of GA-dimerized proteins. The dissociation constant of GA has been estimated to be 10 to 100 times lower than that of ABA38,39. We also quantified dose responses for the four activation systems by measuring EGFP after inducing with varying concentrations of ABA or GA (Fig. 2c,d). We observed wide linear ranges of ABA and GA responsiveness, implying that dosage can modulate activation strength. At the highest tested dose of GA (100 μM), EGFP levels declined (Fig. 2d), possibly owing to a cell acidification effect previously described24.

(a,b) 7-d timecourse for clonal HEK293T pTRE3G–EGFP cells stably expressing Sp or Sa sgTRE3G and (a) ABA-inducible or (b) GA-inducible VPR–Sp or Sa dCas9. Cells were continuously induced with 100 μM ABA or 10 μM GA for 7 d (ON7); induced for 2 d then cultured without inducer for 5 d (ON2 OFF5); or induced for 2 d, cultured without inducer for 3 d, then reinduced for 2 d (ON2 OFF3 ON2). The data are displayed as individual data points for two independent platings of a stable cell line (n = 2). (c,d) Dose response after 48 h induction for HEK293T pTRE3G–EGFP cells transfected with Sp or Sa sgTRE3G and (c) ABA-inducible or (d) GA-inducible VPR–Sp or Sa dCas9. The shaded regions indicate the approximate linear dose–response ranges. The data are displayed as mean ± s.d. for four independent transfections (n = 4).
We additionally characterized the dynamics of the ABA-inducible KRAB–Sp dCas9 repression system. Similar to activation, ABA-induced repression was maintained over 7 d under continuous treatment (Supplementary Fig. 4b). Upon removal of ABA, EGFP continued to decrease for 24 h before recovering. We suspect that this delay was caused by the lingering effects of epigenetic modifications initiated by KRAB40. ABA-inducible repression also demonstrated dose-responsive behavior, as the repressive effect increased at higher doses (Supplementary Fig. 4c). Unlike activation, repression was observed at low doses, which may indicate that various transcriptional effectors require different effective concentrations at the promoter to achieve regulation.
Orthogonal gene regulation by inducible orthogonal dCas9 regulators
The addition of a second, independently controlled dCas9 transcriptional regulator increases the flexibility and scope of dCas9 tools to allow simultaneous regulation of orthogonal genes. We evaluated whether inducible Sa dCas9 could be used alongside Sp dCas9 in the same cell without crosstalk. To achieve complete orthogonality, two criteria must be met: (i) the two inducers must not cross-activate or interfere with each other; and (ii) each of the dCas9s must not interact with the sgRNAs of the other dCas9 (Supplementary Fig. 5a). To address the first criterion, we demonstrated that ABA could not induce heterodimerization of GA-inducible Sp dCas9 and did not interfere with the ability of GA to dimerize GA-inducible Sp dCas9 (Supplementary Fig. 5b). Conversely, GA did not cross-induce or block dimerization of ABA-inducible Sp dCas9. Next, using different combinations of dCas9 activators and sgRNAs, we showed that gene activation only occurred when Sp dCas9 was expressed with Sp sgRNA or Sa dCas9 was expressed with Sa sgRNA, suggesting the absence of crosstalk between dCas9 orthologs and sgRNAs (Supplementary Fig. 5c,d).
We then used the constitutive, direct fusion forms of KRAB–Sp dCas9 and VPR–Sa dCas9 to demonstrate simultaneous regulation of two genes, one activated and one repressed. We expressed Sp and/or Sa dCas9 constructs in a HEK293T pSV40–EGFP pTRE3G–mCherry dual reporter cell line along with Sp sgSV40 and/or Sa sgTRE3G (Fig. 3a). Cells only expressing KRAB–Sp dCas9 and Sp sgSV40 repressed EGFP, while cells only expressing VPR–Sa dCas9 and Sa sgTRE3G activated mCherry. Cells coexpressing both sets of dCas9s and sgRNAs concurrently repressed EGFP and activated mCherry.

(a,b) Fluorescence quantifications for HEK293T pSV40–EGFP pTRE3G-mCherry dual reporter cells transfected with (a) direct fusion KRAB–Sp dCas9 and/or VPR–Sa dCas9 plus their respective sgRNAs or (b) ABA-inducible KRAB–Sp dCas9, GA-inducible VPR–Sa dCas9, and their respective sgRNAs. EGFP fluorescence is displayed for quantifications performed after 5 d induction, and mCherry fluorescence is displayed for quantifications performed after 48 h induction. Fold-change activation is indicated in black, while inverse fold-change repression is indicated in magenta. The data represent four independent transfections performed in technical replicates (n = 8). Dots represent individual data points. The different colors represent different treatment conditions: gray, no inducer; yellow, ABA; green, GA; blue, both ABA and GA. (c,d) Immunofluorescence quantifications for CXCR4 and CD95 after 48 h induction for HEK293T cells transiently transfected with (c) direct fusion KRAB–Sp dCas9, VPR–Sa dCas9, and sgRNAs or (d) GA-inducible VPR–Sp dCas9, ABA-inducible VPR–Sa dCas9, and sgRNAs and stained with APC-conjugated CXCR4 and PE-conjugated CD95 antibodies. Transfected sgRNAs consisted of three targeting sgRNAs (Sp sgCD95-1 to 3 or Sa sgCXCR4-1 to 3) per gene of interest or an equimolar amount of nontargeting sgRNA (Sp/Sa sgTRE3G). The data represent four independent transfections (n = 4). P values from Games–Howell post hoc tests following Welch's ANOVA are provided in Supplementary Table 1.
Finally, we cointroduced ABA-inducible KRAB–Sp dCas9 and GA-inducible VPR–Sa dCas9 into dual reporter cells along with Sp sgSV40 and Sa sgTRE3G (Fig. 3b). We observed that cells only induced with ABA repressed EGFP, and cells only induced with GA activated mCherry. Cells given both inducers simultaneously activated mCherry and repressed EGFP. Importantly, 2D flow cytometry plots show that under simultaneous ABA and GA induction, the subpopulation of cells with increased mCherry also exhibited decreased EGFP, confirming that the same cells are responding to both inducers (Supplementary Fig. 6a).
Orthogonal and inducible endogenous gene activation
We further showed that the dCas9 orthologs can independently regulate expression of discrete endogenous gene targets. In HEK293T cells, direct fusion VPR–Sp dCas9 activated CD95 expression, and VPR–Sa dCas9 concurrently activated CXCR4 expression when coexpressed with their respective sgRNAs (Fig. 3c). When GA-inducible VPR–Sp dCas9 and ABA-inducible VPR–Sa dCas9 were coexpressed instead, GA and ABA could independently trigger activation of CD95 and CXCR4 expression, respectively (Fig. 3d). 2D flow cytometry plots show that under induction by both GA and ABA, individual cells simultaneously expressed higher levels of CD95 and CXCR4 (Supplementary Fig. 6b). Thus, these results demonstrate that two inducible dCas9 transcriptional regulators can act in one cell in an orthogonal fashion.
Coupling multiple chemical-inducible domains for Boolean logic operations
We then looked to expand the vertical flexibility of dCas9 regulators by adding additional layers of inducible control to a single dCas9. Given the robustness of the ABA- and GA-inducible systems, we combined them to generate transcriptional Boolean logic gates. We first created an OR gate by fusing the GA-inducible GAI domain onto the N terminus and the ABA-inducible ABI domain onto the C terminus of Sp dCas9 (Fig. 4a). When coexpressing this dCas9 protein with both GID1–VPR and PYL1–VPR, HEK293T pTRE3G–EGFP cells showed strong EGFP activation (76- to 139-fold) upon induction with ABA alone, GA alone, or both ABA and GA (Fig. 4a).

(a,b) Fluorescence quantifications after 48 h induction for HEK293T pTRE3G–EGFP cells transiently transfected with Sp sgTRE3G and (a) the OR gate or (b) the AND gate VPR–Sp dCas9 construct. The dotted line represents the mean EGFP fluorescence in uninduced cells. The data represent four independent transfections performed in technical replicates (n = 8). (c) Schematic representation of inducibly recruiting opposing transcriptional effectors to a single dCas9. (d) Fluorescence quantifications after 5 d induction for HEK293T pSV40–EGFP cells transfected with the diametric Sp dCas9 construct and Sp sgSV40. Fold-change activation is indicated in black, while inverse fold-change repression is indicated in magenta. The data represent two independent transfections performed in technical replicates (n = 4). (e) 7-d inducer switch timecourse of HEK293T pSV40–EGFP cells stably expressing the diametric Sp dCas9 construct and Sp sgSV40. Cells were induced 2 d with 10 μM GA then replaced with 100 μM ABA for 5 d. Alternatively, cells were induced 5 d with 100 μM ABA then replaced with 10 μM GA for 2 d. The dotted line represents the mean EGFP fluorescence in uninduced cells. The data represent individual data points for two independent platings of a stable cell line (n = 2). P values from Games–Howell post hoc tests following Welch's ANOVA are provided in Supplementary Table 1.
We next sought to generate an AND gate by serially linking two heterodimerization domains (Fig. 4b). The GA-inducible GAI domain was fused to dCas9, the ABA-inducible PYL1 domain to VPR, and the ABI domain to GID1. While cells coexpressing these constructs showed no increase in EGFP when treated with either inducer alone, simultaneous treatment with both inducers produced a 49-fold increase in EGFP (Fig. 4b). We also exchanged the positions of the ABA-inducible and GA-inducible domains and found retention of AND gate function (Supplementary Fig. 7a). Furthermore, we used analogous designs to successfully generate NOR and NAND gates by replacing VPR with KRAB, though NAND showed modest repression (Supplementary Fig. 7b,c). Our results demonstrate an expandable, modular framework for integrating two or more signals into a single dCas9 regulator for logic-based control of gene expression.
Implementing a dual-input inducible dCas9 for diametric gene regulation
Diametric upregulation and downregulation of genes provides a flexible approach for functional interrogation by extending the dynamic range of expression. We created a diametric dCas9 system induced by different inputs to alternatively recruit opposing transcriptional effectors that activate or repress gene expression. This system utilizes the dCas9 from the OR gate coexpressed with GID1–VPR and PYL1–KRAB (Fig. 4c). In pSV40–EGFP cells, ABA-induced recruitment of KRAB resulted in 2.7-fold EGFP repression, while GA-induced recruitment of VPR resulted in 2.5-fold activation (Fig. 4d). Interestingly, coinducing with both ABA and GA to simultaneously recruit KRAB and VPR also led to 2.5-fold EGFP repression, suggesting that repression from KRAB may be dominant to activation from VPR. We further characterized this system's ability to switch from an activated to repressed state and vice versa (Fig. 4e). Replacement of either inducer with the other led to an immediate reversal of the transcriptional regulatory effect, and EGFP levels subsequently passed the basal expression level within 48 h. Compared to simple removal of an inducer from individual activator or repressor systems (Fig. 2b and Supplementary Fig. 4b), recruiting an opposing effector accelerated reversal of the initial regulatory effect.
Discussion
Using the ABA and GA chemical-inducible systems, we were able to achieve strong inducible activation of gene expression. Unlike inducible promoter systems and previously generated inducible dCas9s, we detected no leaky activity from our inducible dCas9s. The inducible dCas9s functioned at levels comparable to direct-fusion dCas9 activators, with added features for reversible and dose-dependent gene regulation. For repression, KRAB–dCas9 acts additively via dCas9 steric hindrance of RNA polymerase and via KRAB-induced repressive chromatin modifications12,13,40. In our dimerization strategy, since dCas9 remains DNA bound in the uninduced state, achieving inducible repression relies on minimizing the steric effect while maximizing the chromatin modification effect. This outcome was achieved by constraining sgRNA binding sites to promoter regions upstream of the transcription start site (TSS). Alternative approaches for inducible repression include using a split dCas9 (refs. 20 and 41) or a caging mechanism42.
Orthogonal dCas9 systems allow simultaneous activation and repression of different genes in one cell. By further pairing each dCas9 with a discrete inducible system, we enable independent temporal control of each gene target. Inducible and orthogonal dCas9-mediated regulation of discrete genes at different timepoints provides new means to dissect the causal factors that govern complex and dynamic processes such as cell differentiation and oncogenesis (Fig. 5). The orthogonal dCas9s also immediately permit functional genomic screens based on combinatorial activation and repression, currently limited to activation or repression only. Furthermore, additional dCas9 orthologs or nuclease-dead mutants of the type V CRISPR protein family Cpf1 (ref. 43) can be integrated into our platform to increase the sets of genes simultaneously manipulated.

Inducible dCas9 regulators can be used in combination in cells to achieve orthogonal and multiplexed transcriptome manipulation.
Engineering additional layers of control for individual dCas9 regulators via Boolean logic gates can be useful when manipulating natural gene networks. For example, in the context of gene-based therapeutics, Boolean logic has been used to increase specificity when rewiring natural gene circuits to produce novel functional outputs44,45. Parallel fusion of dimerization domains also enables recruitment of disparate effectors to the same site, as shown by diametric gene regulation. Expanding the method of parallel effector recruitment to other synergistic transcriptional or epigenetic regulators46,47 may allow inducible encoding of combinatorial regulatory functions at a target locus and enable fine-tuned control of both magnitude and permanence of target gene expression.
Several proof-of-principle studies have demonstrated that dCas9-based synthetic gene circuits can operate in mammalian cells34,48,49. In these studies, synthetic circuit regulation was achieved by conditionally expressing sgRNAs from engineered Pol II promoters. A proposed alternative approach is to modulate the activity of dCas9 itself. However, the key challenge in this strategy is developing multiple dCas9 regulators that can act without crosstalk and can be independently regulated21. The ABA- and GA-inducible Sp and Sa dCas9 systems developed in this work satisfy both requirements. The Boolean logic gates generated here also offer a protein-based alternative for processing multiple inputs to produce a functional output.
Synthetic biology aims to build complex biological systems from simple biological parts. In this study, we demonstrate a synthetic biology approach for creating a modular dCas9 system that responds to chemical inputs and produces disparate or orthogonal transcriptional outputs. We envision that our system can be expanded into a 'plug-and-play' toolbox to generate a vast array of increasingly complex dCas9 transcriptional regulators, and that it will facilitate engineering the transcriptome to elucidate causal relationships between gene networks and cellular phenotypes.
Methods
Step-by-step protocol.
A step-by-step protocol for dual activation of CD95 and CXCR4 in HEK293T cells using orthogonal inducible dCas9s has been included as a Supplementary Protocol and is available at Protocol Exchange (http://dx.doi.org/10.1038/protex.2016.080).
Plasmid design and construction of inducible dCas9s.
Individual constructs for dCas9s, effectors, and sgRNAs used in this study are described in Supplementary Table 2, ordered by figure. Protein domain peptide sequences and sgRNA nucleotide sequences are included in Supplementary Notes 1 and 2 (plasmids no. 84239–84261). New constructs from this study are available on Addgene.
dCas9 fusions. Human codon-optimized Streptococcus pyogenes dCas9 with two C-terminal SV40 nuclear localization signals (NLSs) or Staphylococcus aureus dCas9 with an N-terminal SV40 NLS and C-terminal nucleoplasmin NLS (gift from Feng Zhang, MIT, Addgene plasmid no. 61594)32 was fused at the N terminus to a heterodimerization domain and tagBFP. For the OR gate and diametric dCas9, a second heterodimerization domain (ABI) was fused to the C terminus of dCas9. Expression of dCas9 was driven by a PGK promoter.
Transcriptional effector fusions. The VPR activator22 was assembled by fusing the activation domain of VP64 with the activation domains of p65 (p65AD) and RTA via two GS linkers. An extra SV40 NLS was inserted between VP64 and p65AD. The tripartite sequence was then fused onto the C terminus of a heterodimerization domain. The VP64 activator, KRAB repressor, and GCN4 SunTag scaffold were fused to the C terminus of the heterodimerization domain in an analogous fashion. Expression of the transcriptional effector was driven by a CAG promoter.
Heterodimerization domains. The following heterodimerization domains were used: ABI–PYL1 (gift from J. Crabtree, Stanford, Addgene plasmid no. 38247)23, GAI–GID1 (gift from T. Inoue, Johns Hopkins, Addgene plasmid nos. 37309, 37305)24, FKBP–FRB (gift from T. Meyer, Stanford, Addgene plasmid nos. 20160, 20148)25, PhyB–PIF and CRY2PHR–CIBN (gift from C. Tucker, U of Colorado, Addgene plasmid nos. 26866, 26889)27, and GI–FKF1 (gift from R. Dolmetsch, Stanford, Addgene plasmid nos. 42499, 42500)28.
Transcriptional effectors and dCas9s were amplified by PCR and cloned onto a PiggyBac vector by Gibson assembly. An IRES-driven puromycin- or zeocin-resistance gene and a WPRE sequence were placed 3′ of the transcriptional effector and 5′ of the dCas9. For the AND gate, the IRES-puro was replaced by a p2A–GID1–ABI fusion. Variants were generated by restriction digest and InFusion (Clontech) cloning.
The sgRNAs were cloned onto a pHR lentiviral U6-based expression vector that coexpressed mCherry, EGFP, or mIFP (infrared fluorescent protein, gift from X. Shu, UCSF) from a CMV promoter. For the OR gate and diametric dCas9, the CMV promoter was used to drive expression of the second heterodimerizer–transcriptional effector cassette followed by an IRES–mCherry. Alternative sgRNA sequences were generated by PCR and introduced by InFusion cloning into a vector digested with BstXI and NotI (New England BioLabs).
Construct optimization.
A series of different construct designs using various promoters, heterodimer domain configurations, and plasmid backbones were tested to optimize the activity of the inducible dCas9s (Supplementary Fig. 3 and unpublished data). We noted that the maximal level of activation was determined to occur at high expression levels of the transcriptional effector and low expression levels of the dCas9. To increase expression of the transcriptional effector, a strong constitutive promoter (pCAG) was used to drive transcription, and a WPRE sequence was added to enhance expression. Adding an SV40 polyA transcriptional terminator after the WPRE was found to have disparate effects on different constructs, and the empirically determined best construct was selected in each case. To reduce expression of dCas9, a weak constitutive promoter (pPGK) was selected.
Cell culture and reporter cell line generation.
HEK293T cells (Clontech no. 632180) were maintained in Dulbecco's modified Eagle medium (DMEM) containing 10% FBS, 25 mM D-glucose, and 1.0 mM sodium pyruvate (Life Technologies) in incubators at 37 °C and 5% CO2. Cells were not tested for authenticity and were not routinely tested for mycoplasma contamination. The HEK293T pTRE3G–EGFP and HEK293T pSV40–EGFP reporter lines were generated by transducing cells with lentivirus containing EGFP expressed from a pTRE3G (Clontech) or pSV40 promoter. The HEK293T pSV40–EGFP pTRE3G–mCherry cell line was generated by transducing HEK293T pSV40–EGFP reporter cells with lentivirus expressing mCherry from a pTRE3G promoter. Pure populations of reporter cells were bulk sorted by fluorescence activated cell sorting (FACS) using a BD FACS Aria2 for stable fluorescent marker expression. In the case of HEK293T pTRE3G–EGFP, 1 μg/mL doxycycline was added, and cells were transfected with a transactivator rtTA plasmid (Clontech) for sorting of EGFP+ reporter cells, which were then cultured in the absence of doxycycline after sorting.
Lentivirus production.
Lentivirus was generated by transiently transfecting HEK293T cells with the pHR construct of interest, pCMV–dR8.91, and pMD2.G at a ratio of 9:8:1, respectively. Cell media was changed 24 h after transfection. Viral supernatant was collected 48 h post-transfection, passed through a 0.45 μm filter, and concentrated 10× using the Lenti-X Concentrator (Clontech) by centrifugation at 4 °C for 45 min at 1,500× g following incubation overnight at 4 °C.
Stable dCas9 cell line generation.
To generate cell lines stably expressing PiggyBac-based dCas9 and sgRNA constructs, reporter cells were seeded at a density of 2 × 105 per well in a 12-well plate and transfected the following day with 0.5 μg PiggyBac plasmid containing dCas9 and 0.2 μg Super PiggyBac Transposase plasmid (Systems Biosciences). Approximately 10 d after transfection, cells were sorted by FACS using a BD FACS Aria2 for stable fluorescent marker expression. Where necessary, cells were transduced with lentivirus containing sgRNA before FACS.
Induction experiments.
HEK293T cells were seeded at a density of 3 × 104 per well in 24-well plates (reporter) or 6 × 104 per well in 12-well plates (endogenous gene). For transient transfection experiments, cells were transfected 1 d after seeding with 0.5 μg total of dCas9 and 0.5 μg total of sgRNA per well using TransIT-LT1 transfection reagent (Mirus) at a ratio of 3 μL transfection reagent per 1 μg plasmid. Transfected cells were induced 1 d after transfection by treatment with 100 μM abscisic acid (ABA, Sigma), 10 μM gibberellic acid acetoxymethyl ester (GA, Santa Cruz), 1 μM rapamycin analog (A/C Heterodimerizer, Clontech), or they were placed in light. For red light induction, 15 μM phycocyanobilin (Santa Cruz) was added to cells 3 h before the start of induction, and 10 μW 650 nm light was pulsed in cycles of 20 s ON, 60 s OFF. For blue light, 50 μW 450 nm light was pulsed in cycles of 500 ms ON, 5 s OFF. Stable cell lines were instead induced 1 day after seeding.
Cells were collected after 48 h induction for activation experiments, after 5 d induction for repression experiments, or after 7 d induction for timecourse experiments. For repression experiments, cells were passaged after 48 h induction and treated with fresh inducer. For timecourse experiments, cells were passaged after 48 h and 5 d induction and treated with fresh inducer. To remove inducer during timecourses, inducer-treated media was aspirated and replaced with fresh media. No additional washing was performed when removing inducer to minimize detachment and loss of low-confluence cells.
Fluorescence and immunostaining assays using flow cytometry analysis.
To analyze fluorescent protein expression, cells were dissociated using 0.05% Trypsin EDTA (Life Technologies) and analyzed by flow cytometry on a BD LSRII. To analyze surface marker expression, cells were instead dissociated using the nonenzymatic reagent Versene (Life Technologies), then stained in 10% FBS in PBS for 1 h at room temperature. Antibodies were purchased from BioLegend (nos. 306510, 400322, 305607, 400111). CXCR4 antibody and isotype were used at 0.4 μg/mL, and CD95 antibody and isotype were used at 0.5 μg/mL.
Flow cytometry data was analyzed using FlowJo. At least 10,000 cells were analyzed for each sample. For stably integrated cell lines, cells were gated for positive fluorescent marker expression corresponding to construct expression. For 48 h transfection experiments, cells were gated for positive dCas9 (tagBFP) expression, then further gated for the highest 50% of sgRNA (mCherry, EGFP, or mIFP) expressing cells. For 5 d transfection experiments, cells were gated for the highest 20% of sgRNA expression on account of dilution of transfected plasmids. The mean intensity of the reporter fluorescent protein (EGFP or mCherry) or immunofluorescent antibody (APC-CXCR4 or PE-CD95) was measured. When calculating the fold change of endogenous proteins, we subtracted the fluorescence level of isotype-stained wild-type HEK293T cells from the measured fluorescence of samples.
Experimental design and statistics.
Data are displayed as individual points or as mean ± s.d., with sample size indicated in figure legends. No sample size estimates were performed, and the sample sizes used in this study are consistent with those used by similar genome editing and gene regulation studies. No samples were excluded from analysis. No randomization or blinding was performed.
Statistical analysis was performed using SPSS Statistics 21 (IBM). Equal variance between populations was not assumed. To account for unequal variance among conditions, Welch's two-sided t-test was performed when comparing two conditions, and Welch's ANOVA was performed followed by Games–Howell post hoc tests when comparing more than two conditions with each other.
References
Maurano, M.T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Weinhold, N., Jacobsen, A., Schultz, N., Sander, C. & Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 46, 1160–1165 (2014).
DeRisi, J.L., Iyer, V.R. & Brown, P.O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278, 680–686 (1997).
Nagalakshmi, U. et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349 (2008).
Loew, R., Heinz, N., Hampf, M., Bujard, H. & Gossen, M. Improved Tet-responsive promoters with minimized background expression. BMC Biotechnol 10, 81 (2010).
No, D., Yao, T.P. & Evans, R.M. Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proc. Natl. Acad. Sci. USA 93, 3346–3351 (1996).
Doudna, J.A. & Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096 (2014).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Jiang, W., Bikard, D., Cox, D., Zhang, F. & Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239 (2013).
Qi, L.S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Gilbert, L.A. et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 (2013).
Maeder, M.L. et al. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 10, 977–979 (2013).
Perez-Pinera, P. et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 10, 973–976 (2013).
Mali, P. et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838 (2013).
Cheng, A.W. et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 23, 1163–1171 (2013).
Polstein, L.R. & Gersbach, C.A. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat. Chem. Biol. 11, 198–200 (2015).
Nihongaki, Y., Yamamoto, S., Kawano, F., Suzuki, H. & Sato, M. CRISPR-Cas9-based photoactivatable transcription system. Chem. Biol. 22, 169–174 (2015).
Zetsche, B., Volz, S.E. & Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 33, 139–142 (2015).
Jusiak, B., Cleto, S., Perez-Piñera, P. & Lu, T.K. Engineering synthetic gene circuits in living cells with CRISPR technology. Trends Biotechnol. 34, 535–547 (2016).
Chavez, A. et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 12, 326–328 (2015).
Liang, F.-S., Ho, W.Q. & Crabtree, G.R. Engineering the ABA plant stress pathway for regulation of induced proximity. Sci. Signal. 4, rs2 (2011).
Miyamoto, T. et al. Rapid and orthogonal logic gating with a gibberellin-induced dimerization system. Nat. Chem. Biol. 8, 465–470 (2012).
Inoue, T., Heo, W.D., Grimley, J.S., Wandless, T.J. & Meyer, T. An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat. Methods 2, 415–418 (2005).
Levskaya, A., Weiner, O.D., Lim, W.A. & Voigt, C.A. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461, 997–1001 (2009).
Kennedy, M.J. et al. Rapid blue-light-mediated induction of protein interactions in living cells. Nat. Methods 7, 973–975 (2010).
Yazawa, M., Sadaghiani, A.M., Hsueh, B. & Dolmetsch, R.E. Induction of protein-protein interactions in live cells using light. Nat. Biotechnol. 27, 941–945 (2009).
Liu, H. et al. Photoexcited CRY2 interacts with CIB1 to regulate transcription and floral initiation in Arabidopsis. Science 322, 1535–1539 (2008).
Toettcher, J.E., Gong, D., Lim, W.A. & Weiner, O.D. Light control of plasma membrane recruitment using the Phy-PIF system. Methods Enzymol. 497, 409–423 (2011).
Margolin, J.F. et al. Krüppel-associated boxes are potent transcriptional repression domains. Proc. Natl. Acad. Sci. USA 91, 4509–4513 (1994).
Ran, F.A. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015).
Nishimasu, H. et al. Crystal structure of Staphylococcus aureus Cas9. Cell 162, 1113–1126 (2015).
Kiani, S. et al. Cas9 gRNA engineering for genome editing, activation and repression. Nat. Methods 12, 1051–1054 (2015).
Chen, B. et al. Expanding the CRISPR imaging toolset with Staphylococcus aureus Cas9 for simultaneous imaging of multiple genomic loci. Nucleic Acids Res. 44, e75 (2016).
Tanenbaum, M.E., Gilbert, L.A., Qi, L.S., Weissman, J.S. & Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635–646 (2014).
Chavez, A. et al. Comparison of Cas9 activators in multiple species. Nat. Methods 13, 563–567 (2016).
Ueguchi-Tanaka, M. et al. GIBBERELLIN INSENSITIVE DWARF1 encodes a soluble receptor for gibberellin. Nature 437, 693–698 (2005).
Miyazono, K. et al. Structural basis of abscisic acid signalling. Nature 462, 609–614 (2009).
Thakore, P.I. et al. Highly specific epigenome editing by CRISPR–Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149 (2015).
Nihongaki, Y., Kawano, F., Nakajima, T. & Sato, M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 33, 755–760 (2015).
Hemphill, J., Borchardt, E.K., Brown, K., Asokan, A. & Deiters, A. Optical Control of CRISPR/Cas9 Gene Editing. J. Am. Chem. Soc. 137, 5642–5645 (2015).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015).
Roybal, K.T. et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779 (2016).
Morsut, L. et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell 164, 780–791 (2016).
Kearns, N.A. et al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 12, 401–403 (2015).
Hilton, I.B. et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 33, 510–517 (2015).
Kiani, S. et al. CRISPR transcriptional repression devices and layered circuits in mammalian cells. Nat. Methods 11, 723–726 (2014).
Nissim, L., Perli, S.D., Fridkin, A., Perez-Pinera, P. & Lu, T.K. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol. Cell 54, 698–710 (2014).
Acknowledgements
The authors thank the members of the Qi and Lim labs for advice and helpful discussions and the Stanford Shared FACS Facility for technical support. The authors also thank X. Shu (UCSF), F. Zhang (MIT), G. Crabtree (Stanford), T. Inoue (Johns Hopkins), T. Meyer (Stanford), C. Tucker (U of Colorado), and R. Dolmetsch (Stanford) for constructs used in this study. L.S.Q. acknowledges support from the NIH Office of the Director (OD) and the National Institute of Dental & Craniofacial Research (NIDCR). Y.G. acknowledges support from the Stanford Cancer Biology Graduate Program and the NSF GRFP fellowship. X.X. acknowledges the support from the Helen Hay Whitney Foundation postdoctoral fellowship. This work was supported by DP5 OD017887 (L.S.Q.), NIH R01 DA036858 (L.S.Q. and W.A.L.), NIH P50 GM081879 (W.A.L.), and the Howard Hughes Medical Institute (W.A.L.).
Author information
Authors and Affiliations
Contributions
L.S.Q. and W.A.L. conceived of the research; Y.G. and X.X. designed the study; Y.G., X.X., S.W., and E.J.C. performed the experiments; Y.G., X.X., S.W., and L.S.Q. analyzed the data; Y.G., X.X., and L.S.Q. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Screen of chemical- and light-mediated heterodimerization systems for inducible dCas9 gene activation.
(a) PiggyBac single vector design for expression of dCas9 and effector cassettes. (b) Binding sites of Sp sgTRE3G on the pTRE3G promoter. (c-f) Fluorescence quantification after 48 h induction for HEK293T pTRE3G-EGFP cells transfected with Sp TRE3G and (c) rapamycin-inducible, (d) PHYB/PIF red light-inducible, (e) CRY2PHR/CIBN blue light inducible, or (f) FKF1/GI blue light-inducible VPR-Sp dCas9. The data are displayed as mean ± SD for two (d-f) or four (c) independent transfections performed in technical replicates (n = 4 or 8). (* P < 0.05, ** P < 0.01, *** P < 0.001, n.s. = not significant, Games-Howell post-hoc test following Welch’s ANOVA).
Supplementary Figure 2 Gene activation and repression by direct fusion dCas9s.
(a) Fluorescence quantification for HEK293T pTRE3G-EGFP cells 3 days following transfection with Sp sgTRE3G and VPR-Sp dCas9. (b) Fluorescence quantification for HEK293T pSV40-EGFP cells 6 days following transfection with Sp sgSV40 and KRAB-Sp dCas9. (c) Fluorescence quantification for HEK293T pTRE3G-EGFP cells 3 days following transfection with Sa sgTRE3G and VPR-Sa dCas9. Fold-change activation is indicated in black, while inverse fold-change repression is indicated in magenta. The data are displayed as mean ± SD for four independent transfections performed in technical replicates (n = 8). (* P < 0.05, ** P < 0.01, *** P < 0.001, n.s. = not significant, Welch’s two-sided t test).
Supplementary Figure 3 Gene activation by different configurations of inducible dCas9s.
(a-c) Fluorescence quantification after 48 h induction for HEK293T pTRE3G-EGFP cells stably expressing Sp sgTRE3G and one of two configurations of (a) ABA-inducible, (b) PHYB/PIF red light-inducible, or (c) rapamycin-inducible VP64-Sp dCas9. The data are displayed as mean ± SD for two independent platings of a stable cell line performed in technical replicates (n = 4). †Data collected from a separate experiment consisting of one plating. (d-f) Fluorescence quantification after 48 h induction for HEK293T pTRE3G-EGFP cells transfected with Sp sgTRE3G and one of two configurations of (d) GA-inducible, (e) CRY2PHR/CIBN blue light-inducible, or (f) FKF1/GI blue light-inducible VP64-Sp dCas9. The data are displayed as mean ± SD for two independent transfections performed in technical replicates (n = 4).
Supplementary Figure 4 Gene repression characterization for ABA-inducible KRAB-Sp dCas9.
(a) Binding sites of Sp sgSV40 on the pSV40 promoter. (b) 7 d timecourse for clonal HEK293T pSV40-EGFP cells stably expressing Sp sgSV40 and ABA-inducible KRAB-Sp dCas9. Cells were induced with ABA for 7 d (ON7) or for 4 and 5 d followed by ABA removal (ON4 OFF3 and ON5 OFF2). (c) ABA dose response after 5 d induction for clonal HEK293T pSV40-EGFP cells stably expressing Sp sgSV40 and ABA-inducible KRAB-Sp dCas9. The data are displayed as mean ± SD for two (b) or four (c) independent platings of a stable cell line (n = 2 or 4).
Supplementary Figure 5 Orthogonality of inducible dCas9 components.
(a) Fully orthogonal inducible dCas9 systems require each component of one system to avoid crosstalk to its counterpart in the paired system. (b) Fluorescence quantification after 48 h induction of HEK293T pTRE3G-EGFP cells transfected with Sp sgTRE3G and ABA-inducible or GA-inducible VP64-Sp dCas9. (c) Fluorescence quantification for HEK293T pTRE3G-EGFP cells 72 h following transfection with Sp/Sa sgTRE3G and direct fusion VPR-Sp/Sa dCas9. (d) Fluorescence quantification after 48 h induction of HEK293T pTRE3G-EGFP cells transfected with Sp/Sa sgTRE3G and ABA-inducible VP64-Sp/Sa dCas9. The data are displayed as mean ± SD for two independent transfections performed in technical replicates (n = 4). (* P < 0.05, ** P < 0.01, *** P < 0.001, n.s. = not significant, Games-Howell post-hoc test following Welch’s ANOVA).
Supplementary Figure 6 Independent gene regulation by orthogonal dCas9s.
(a) 2-D flow cytometry contour plots of EGFP and mCherry fluorescence quantification after 5 d induction for experiment shown in Fig. 3b. The plots display data from one of four independent transfections for each condition. (b) 2-D flow cytometry contour plots of CD95 and CXCR4 immunofluorescence quantification after 48 h induction for experiment shown in Fig. 3d. The plots display data from one of four independent transfections for each condition.
Supplementary Figure 7 Two-input logic gates for inducible gene activation and repression.
(a) Fluorescence quantification after 48 h induction for HEK293T pTRE3G-EGFP cells transfected with Sp sgTRE3G and one of two configurations of an AND gate VP64-Sp dCas9. (b-c) Fluorescence quantifications after 5 d induction for HEK293T pSV40-EGFP cells transiently transfected with Sp sgSV40 and (a) the NOR gate or (b) the NAND gate KRAB-Sp dCas9 construct. Fold-change activation is indicated in black, while inverse fold-change repression is indicated in magenta. The data are displayed as mean ± SD for two (a, c) or four (b) independent transfections performed in technical replicates (n = 4 or 8). (* P < 0.05, ** P < 0.01, *** P < 0.001, n.s. = not significant, Games-Howell post-hoc test following Welch’s ANOVA).
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–7, Supplementary Tables 1 and 2, Supplementary Notes 1 and 2, and Supplementary Protocol. (PDF 1644 kb)
Rights and permissions
About this article

Cite this article
Gao, Y., Xiong, X., Wong, S. et al. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat Methods 13, 1043–1049 (2016). https://doi.org/10.1038/nmeth.4042
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.4042
This article is cited by
-
Orthogonal inducible control of Cas13 circuits enables programmable RNA regulation in mammalian cells
Nature Communications (2024)
-
A universal deep-learning model for zinc finger design enables transcription factor reprogramming
Nature Biotechnology (2023)
-
Engineering cell-derived extracellular vesicles for gene therapy
Nature Biomedical Engineering (2023)
-
CasTuner is a degron and CRISPR/Cas-based toolkit for analog tuning of endogenous gene expression
Nature Communications (2023)
-
Combinatorial protein dimerization enables precise multi-input synthetic computations
Nature Chemical Biology (2023)
