
Abstract
Magnetotactic bacteria align along the Earth’s magnetic field using an organelle called the magnetosome, a biomineralized magnetite (Fe(ii)Fe(iii)2O4) or greigite (Fe(ii)Fe(iii)2S4) crystal embedded in a lipid vesicle. Although the need for both iron(ii) and iron(iii) is clear, little is known about the biological mechanisms controlling their ratio1. Here we present the structure of the magnetosome-associated protein MamP and find that it is built on a unique arrangement of a self-plugged PDZ domain fused to two magnetochrome domains, defining a new class of c-type cytochrome exclusively found in magnetotactic bacteria. Mutational analysis, enzyme kinetics, co-crystallization with iron(ii) and an in vitro MamP-assisted magnetite production assay establish MamP as an iron oxidase that contributes to the formation of iron(iii) ferrihydrite eventually required for magnetite crystal growth in vivo. These results demonstrate the molecular mechanisms of iron management taking place inside the magnetosome and highlight the role of magnetochrome in iron biomineralization.
Similar content being viewed by others


Main
Magnetotactic bacteria (MTB) have the particular ability to align with geomagnetic field lines, a phenomenon referred to as magnetotaxis. This magnetotactic property is due to the presence of the magnetosome, an organelle made of a lipid vesicle loaded with a single magnetite or greigite crystal about 50 nm in size. The alignment of magnetosomes inside the cell acts like a compass needle to orient MTB passively in geomagnetic fields, putatively simplifying their search for preferred microaerophilic environments. Formation of this iron-rich organelle is genetically orchestrated by genes located in the magnetosome genetic island. These genes ensure the formation of the vesicles, their alignment, their loading with iron and the biomineralization into magnetite or greigite2,3. Despite early observations of redox control in MTB4,5, this last step remains poorly understood, notably the management of the iron(ii) and iron(iii) species required for magnetite or greigite formation. There is indication that some oxidized iron species such as ferrihydrite accumulate before magnetite formation, therefore suggesting the need for a reductive process6,7. However, there is also growing evidence that the readily available iron species in the magnetosome is iron(ii). Both the presence of numerous and active ferric reductases in MTB8 and the predominance of cation diffusion facilitators for iron(ii) trafficking9 associated with the magnetosome support this premise.
The search for potential redox proteins within the magnetosome genetic island has led to the identification of four gene products containing at least two tandem c-type cytochrome motifs CX2CH, recently called magnetochrome domains10: MamE, MamP, MamT and MamX. Among these magnetochrome-containing proteins, MamE and MamP are conserved in all MTB and, interestingly, deletion mutants of the corresponding genes show defects in the biocrystallization process11,12. However, the multiplicity of this domain leads to difficulties in the phenotypic analyses of a single-domain deletion mutant in the mamE gene13, suggesting that magnetochrome domains could be functionally redundant. This magnetochrome domain seems specific to MTB as it has not been found in any other species so far, suggesting it may represent a new functional class of cytochrome.
We purified and crystallized the soluble part of MamP from the MO-1 strain (residues 26–260, see Methods). The MamP structure was subsequently solved by multi-wavelength anomalous diffraction (MAD) using the four iron atoms present in the asymmetric unit (see Extended Data Table 1 for statistics and Extended Data Fig. 1 for an example of the 2mFobs − DFcalc map). The first visible residue in the electron density corresponds to residue 87, indicating the presence of a long flexible arm connecting the protein to the single transmembrane helix. Following this flexible arm, the protein folds as a PDZ domain, a small c-type cytochrome domain (the first magnetochrome domain, MCR1), a 17-residue linker and finally a second magnetochrome domain (Fig. 1; see Extended Data Fig. 2 for an annotated sequence alignment). The first magnetochrome domain is in contact with its own PDZ domain, whereas the second is projected above the PDZ domain of the other monomer. The minimal unit of MamP is a dimer, although the crystal packing could also support the existence of a tetramer made by two symmetric dimers (one in an ‘open’ state and the other in a ‘closed’ state; Extended Data Figs 3 and 4) differing by only small but functionally important side-chain reorientations, as outlined below. In solution, size exclusion chromatography indicates a pH-dependent tetramer/dimer equilibrium that was confirmed by small-angle X-ray scattering (SAXS) whereas circular dichroism measurements indicated no major structural rearrangement upon pH change (Methods and Extended Data Fig. 3).

a, Representation of MamP domain organization. The linker between the transmembrane helix and the PDZ domain is shown as a dashed line, indicating that it is disordered in the crystal structure. b, Three-dimensional structure of MamP with one monomer coloured in grey and the other monomer coloured with a ramp from blue (amino (N) terminus) to red (carboxy (C) terminus).
Using the PDZ domain of MamP, a structural homology search using the DALI server14 indicated that the closest structural homologues are the PDZ domains found in the high-temperature requirement A (HtrA) family of Ser proteases. HtrA proteases combine a protease domain to one or two PDZ domains and are involved in protein quality control15,16. These functions are made possible by the peptide-binding properties of PDZ domains, a domain that folds as a single β-sheet capped on one side by an α-helix, thereby delineating a groove dedicated to peptide substrates by β-strand augmentation17.
Interestingly, the PDZ domain of MamP is unusual because its groove is not open for protein partner binding. Instead, the first visible strand in the MamP structure (β1, denoted as SP for ‘self-plugging’ strand) fills the binding groove found in classical PDZ domains. In MamP, this SP strand clearly contributes to its dimerization (Fig. 2a). Indeed, the position of the SP strand further allows the extension of the β-sheet, with strand β7 connecting the PDZ domain to the first magnetochrome domain. This last strand (denoted as Dim for dimerization strand) largely contributes to the dimeric interface of MamP (Fig. 2a). The overall interface is conserved, suggesting a selective pressure for this oligomeric assembly.

a, The surface representation of the one monomer is colour-coded based on sequence conservation in alignments of all known MamP and indicates that the dimeric interface is conserved. Note that both the self-plugged (SP) and the dimerization (Dim) strands participate in the dimerization of MamP together with strand β3 and helices α1 and α2. b, Superimposition of MCR1 with MCR2 with conserved magnetochrome residues represented in stick.
We recently proposed that the two c-type cytochrome domains of MamP define a new domain exclusively found in MTB10. The present structure determination of MamP allows us to describe the fold of this domain, demonstrating its uniqueness at the structural level. A magnetochrome starts with a hydrophobic residue (ψ1 in Fig. 2b and Extended Data Fig. 2) in direct contact with the haem, followed by a Pro–His (PH) dyad, located five to nine residues upstream of the CXXCH motif, providing the sixth and fifth haem ligands, respectively. Finally, a terminal hydrophobic residue (ψ2 in Fig. 2b) closes the magnetochrome fold through a hydrophobic interaction with the ψ1 residue.
The structure of MamP also confirms that the magnetochrome domain defines a single haem-binding domain belonging to a new family of c-type cytochrome. Indeed, it folds as one of the smallest haem-binding units known thus far, with only 23 residues surrounding the haem (Fig. 2b). This best compares to artificial microperoxidases that possess a covalently attached haem and a single histidine ligand, whereas other mono-haem c-type cytochromes minimally possess about 70 residues surrounding a single haem (Protein Data Bank accession number 1K3G). We found that the haems in both magnetochrome domains are highly solvent exposed, with 281 and 214 Å2 for MCR1 and MCR2, respectively. These values best compare to multihaem cytochromes or proteins with transient affinity for haem such as haemophores or haemopexin18. In addition, the haem-binding mode in magnetochrome domains also stands out because all rings of the haems are solvent exposed, which is not the case in other c-type cytochromes18.
The dimeric structure of MamP creates a large surface-exposed acidic pocket resembling a crucible with approximate dimensions of 8 Å (depth) × 15 Å (diameter) (Fig. 3a). Eight conserved acidic residues from both PDZ domains delineate the bottom of this crucible, whereas the sides are formed by the propionates of the haems and four conserved acidic residues from the linkers between the two magnetochrome domains (Fig. 3a). A conserved histidine residue (H93) is located in the middle of the crucible with a network of conserved hydrogen bonded residues connecting its side chain to the exterior of the protein through polar residues, which is reminiscent of a hydrogen exit channel (Fig. 3b and Extended Data Fig. 5). The peculiar arrangement of conserved acidic residues observed in the MamP cavity suggests the presence of a ‘hot spot’ and led us to investigate how it reacts with iron compounds. In vitro, we found that MamP efficiently oxidized Fe(ii)SO4 at alkaline pH (Fig. 4a). This reaction proceeded with a rate constant (kox) of 1.06 × 10−3 µM–1 s–1 at an optimal pH of 9, which is comparable to observations on the multihaem cytochrome c MtoA, a decahaem c-type cytochrome from Sideroxydans lithotrophicus involved in microbial iron oxidation19. Interestingly, the optimal pH of MamP iron oxidase activity coincides with that found for in vitro magnetite synthesis20.

a, Molecular surface representation of a MamP dimer coloured according to its electrostatic potential. The size of the crucible is indicated. b, Detail of the two acidic networks making the bottom (red circle) and the side (yellow circle) of the crucible.

a, Visible spectra of MamP (5 μM) before (blue line) and after (red line) addition of 50 μM Fe(ii)SO4. Inset: kinetics of reduction of MamP by Fe(ii)SO4 (black dots) and fit of the experimental data points by a single exponential (red line). b, Soaking experiments of MamP crystals with Fe(ii)SO4 at pH 9. The electron density map corresponds to an anomalous map collected at the iron edge and contoured at 5σ all around the dimer. c, Details of the position of the anomalous peak in the conserved acidic pocket. d, e, Genetic complementation studies to examine the function of MamP acidic residues. d, Magnetic response of cells. Data were collected as three biological replicates with two technical replicates per biological replicate. Values are represented as mean ± s.d. (n = 6). e, Representative TEM images of strains (scale bar, 0.2 μm). Cell images are representative of those collected for all three replicates. f, Time-resolved analysis of the mineralization synthesis followed by X-ray diffraction with reference peak of ferrihydrite and magnetite and their relative intensity (the full X-ray diffraction spectrum is shown in Extended Data Fig. 7). The six-line ferrihydrite peak ((112) peak in red dotted line) increases until 50 min whereas the magnetite peak ((400) in green) is only visible after 40 min and increases then, possibly at the expense of the ferrihydrite precursor.
The Fe(ii) oxidation activity detected with MamP in vitro further prompted us to investigate whether this activity could be substantiated by structural approaches and by testing the effect of MamP on magnetite formation in solution. To get an impression of the iron oxidase activity of MamP, we transferred a MamP crystal to pH 9 and then soaked it in a solution containing Fe(ii)SO4 before data collection at the iron edge. The resulting anomalous electron density clearly indicates the presence of an iron-binding site mediated by conserved residues located at the bottom of the crucible (Fig. 4b, c). The anomalous electron density peak is only present in the open dimer and its elongated shape even suggests the presence of a di-iron-binding site with the two iron atoms replacing the two water molecules that are only seen in the high-resolution structure of the open dimer. This stabilization of two iron atoms in the open dimer is in line with the calculated charged states, which suggests that the iron-binding residues are unprotonated in the open dimer (Extended Data Fig. 4). Using a ferrozine assay to estimate the iron/MamP stoichiometry, we found that four irons are oxidized per MamP dimer, which provides a better fit with a di-iron-binding site.
To examine the functional relevance of the conserved acidic residues, some of which are directly involved in iron binding, we used the mamP deletion mutant of Magnetospirillum magneticum AMB-1 (the genetic tools being available for this strain), and complemented this strain with the wild-type gene or a mamP variant in which all conserved acidic residues are mutated to alanine (mamPΔacid). In accordance with our observation of an iron-binding site at the bottom of the crucible, we found that these residues are essential for magnetite formation in vivo as judged by the magnetic response of the cells (Cmag), crystal size distributions as well as transmission electron microscope (TEM) images (Fig. 4d, e and Extended Data Figs 6 and 7).
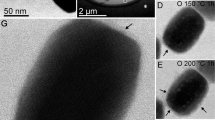
Finally, we observed the role of MamP in a mineralization experiment (Fig. 4f and Extended Data Figs 8 and 9). Indeed, magnetite is typically formed in solution by co-precipitation experiments of iron with the stoichiometric ratio of magnetite (Fe(ii)/Fe(iii) = 0.5) (ref. 21). We decided to start exclusively with Fe(ii) to see if any mineral would form in the presence or absence of MamP. In the presence of MamP, we initially observed the formation of ferrihydrite, an Fe(iii) oxide, with a progressive evolution of this mineral to magnetite (Fig. 4f). The control experiment omitting MamP could not allow the detection of any mineral by X-ray diffraction. TEM images indicated the presence of electron-dense particles that were not seen when MamP was omitted, thereby suggesting MamP-mediated production of ferrihydrite or magnetite (Extended Data Fig. 8). This mechanism is in accord with a process where the Fe(ii) is oxidized by the protein to enable the formation of ferrihydrite, a purely ferric iron oxide. Once MamP is fully reduced, the continuous addition of Fe(ii) enables the transformation of ferrihydrite to magnetite. Such a mechanism strongly resembles the pathway recently described for the synthetic formation of magnetite from solution22.
The structural basis of MamP-mediated iron biomineralization presented here validates an old model put forth for how magnetite formation should be redox-controlled in the magnetosome, and emphasizes the versatility of the magnetochrome domains in this process5. Magnetite crystal growth requires a precise Fe(iii)/Fe(ii) ratio; the MamP properties we showed, both in vitro and in crystallo, would allow the production of the ferrihydrite precursor thanks to the presence of acidic residues at the bottom of a crucible surrounded by four magnetochrome domains. Together with the presence of iron(ii), this ferrihydrite would evolve towards magnetite in the magnetosome. This molecular model fits perfectly with sequential events observed for magnetite biomineralization in MTB6,7. The dimension and acidic nature of the MamP crucible, the presence of a conserved proton exit channels at its bottom and the four haems on either side are well-suited for the expected chemistry of ferrihydrite formation from Fe(ii):

Functional and structural studies are now required to determine the contribution of other magnetochrome-containing proteins in the process of magnetite biomineralization.
Methods Summary
MamP from strain MO-1 was cloned in plasmid pET26b+ and expressed in Escherichia coli. The protein was purified by metal-affinity and gel-filtration chromatography. Protein crystals were obtained at 20 °C, diffraction data were collected at synchrotrons SOLEIL, SLS and ESRF, and the structure was solved by MAD. Iron oxidase activity was measured in an anaerobic glove box. We used strain AMB-1 for in vivo mutational analysis. In vitro biomineralization experiments were done under nitrogenic atmosphere using Fe(ii), and X-ray diffraction was measured at synchrotron BESSY II.
Online Methods
Cloning, protein production and purification
The DNA sequence corresponding to residues D26–Q260 of magnetotactic ovoidal bacterium MO-1 mamP gene was sub-cloned into the plasmid pET26b+ (Novagen) using the In-Fusion PCR Cloning system (Clontech). Amplification was performed with Pfu Ultra High-Fidelity DNA polymerase (New England Biolabs). The resulting clone was co-expressed with the pEC26 (a gift from R. van Lis) clone of the ccm operon in chemically competent E. coli BL21(DE3) cells (Invitrogen).
Bacterial cells were cultured at 37 °C in Super Optimal Broth supplemented with 50 µg ml−1 kanamycin and 50 µg ml−1 chloramphenicol. Expression was induced once cells reach an absorbance of 0.6–0.8 by addition of 20 μM IPTG and left overnight at 37 °C. Cells were collected by centrifugation and re-suspended in lysis buffer (100 mM Na2HPO4, 500 mM NaCl, pH 8.0) supplemented with protease inhibitor cocktail (Sigma-Aldrich) and DNase. Lysis buffer was used in the ratio of 1.5 ml buffer per 1 g of wet cell pellet. Re-suspended pellets were stored at −80 °C.
Cells were disrupted by the One Shot cell disruption system (Constant Systems). Purification of the His-tagged proteins was performed in two steps using Ni-charged HiTrap Chelating HP and HiLoad 16/60 Superdex 200 columns on an ÄKTA FPLC protein purification system (GE Healthcare). Prep Grade columns (GE Healthcare) were pre-equilibrated with IMAC buffer (100 mM HEPES, 500 mM NaCl, pH 7.5) and gel filtration buffer (20 mM HEPES, 200 mM, pH 7.5), respectively. The filtered lysates were loaded onto the HiTrap Chelating columns and washed with IMAC buffer. Bound protein was eluted with IMAC buffer containing 100 mM imidazole and loaded onto the pre-equilibrated gel filtration column. Fractions containing the target proteins were pooled and concentrated using a VIVASPIN centrifugal filter device with a cut-off size of 10 kDa. Protein purity was confirmed by SDS–polyacrylamide gel electrophoresis and protein samples were flash frozen and stored at −80 °C.
Circular dichroism and SAXS measurements
Circular dichroism measurements used MamP (0.1 mg ml−1) in 20 mM phosphate buffer. SAXS measurements were done on beamline SWING at SOLEIL (Paris). This beamline was equipped with a size-exclusion chromatography column (Superdex-200 3.3/30) coupled with ultraviolet detection and followed by online SAX measurements. Molecular masses of the eluted peaks were estimated using the MoW program23.
Crystallization, data collection, structure determination and validation
Native crystals were obtained by the sitting drop vapour diffusion method in a 96-well plate. Half a microlitre of the protein solution (9.7 mg ml−1) in GF buffer was mixed with 0.5 µl of well solution consisting of 0.2 M ammonium nitrate and 20% (w/v) PEG 3350. The plate was incubated at 20 °C and crystals were obtained after 12–14 days. Crystals were quickly transferred to cryogenic solutions containing well solution, 20% ethylene glycol or glycerol, and flash frozen in liquid nitrogen. To obtain structural data on Fe(ii) binding to MamP, a crystal of the protein obtained in 0.2 M ammonium acetate and 20% (w/v) PEG3350 was transferred in the same mother liquor but containing 0.1 M bis–tris propane at pH9. Fe(ii)SO4 was then added to a final concentration of 10 mM for approximately 5 minutes after which the crystal was transferred to a cryogenic condition at pH 9 containing 20% MPD before flash freezing in liquid nitrogen. For the soaking experiments, all the solutions used were de-gassed.
Native data were collected to 1.8 Å on beamline X06SA at Swiss Light Source (Switzerland), whereas MAD data were collected to 2.5 Å on beamline PROXIMA1 at SOLEIL synchrotron facility (Gif-sur-Ivette, France). The crystal soaked in Fe(ii) was collected to 2.8 Å on beamline FIP (ESRF, France). For the native crystals, data integration was performed in XDS and scaled with XSCALE (MAD data)24 or SCALA (native data)25. For the Fe(ii)-soaked crystal, data integration was performed with Mosflm26 and integrated with SCALA. The structure was solved by the autoSHARP program27 using the MAD data set.
All the structures were refined with Refmac5 (ref. 28) with a final refinement cycle using PHENIX29 in the case of the native structure. Structure validations were performed using the Molprobity server (http://molprobity.biochem.duke.edu/) and Procheck from the CCP4 suite.
Solution studies, mineralization experiments and analysis
The iron oxidase properties of MamP were studied in an anaerobic glove box filled with N2 (<10 p.p.m. oxygen). The oxidation state of MamP was monitored by following the absorption of haem at either 417 or 551 nm. The ferrozine assay was also done in the anaerobic glove box. In this case, the diminution of Fe(ii) concentration due to oxidation by MamP was estimated by mixing 400 μL of sample (5 μM MamP in a 50 mM bis–tris propane, 150 mM NaCl buffer, pH 8) with 1.6 ml of Ferrozine (1% w/v in 50 mM HEPES, pH 7). Fe(ii) concentration was estimated by comparison with a standard curve using known Fe(ii) concentrations.
The mineralization experiments were done similarly to the modified co-precipitation method developed by our group20,22. Briefly, the computer-controlled setup consists of a titration device (Metrohm 888 Titrando), a dosing device (Metrohm 805 Dosimat) and a pH electrode (Metrohm Biotrode). The titration device provided a 0.1 M NaOH solution, and in the experiment reported here, the dosing device was filled with a 0.1 M ferrous solution to test the ability of the protein to oxidize iron to its ferric form and to the associated mineral phases. The solutions were prepared from 1 M sodium hydroxide solution (Merck) and ferrous chloride tetrahydrate (Sigma-Aldrich).
Before starting an experiment the original MamP-solution (3.2 ml, 200 µM; Hepes 20 mM, NaCl 500 mM) was dialysed with 200 mM NaCl solution to avoid the presence of foreign ions in the synthesis. All experiments were done at room temperature and under nitrogenic atmosphere, the latter to avoid possible oxidation by air. All the solutions were carefully purged with nitrogen before use.
The reactor vessel was then filled with the dialysed MamP solution (14 ml, 22.9 µM) which was de-gassed for 30 min. Alternatively, the reaction vessel was filled with de-ionized water as a control. The pH was adjusted to 9 or 10 respectively before the crystallization experiment started. The pH was kept constant during the synthesis with the help of the titration device. The synthesis was performed by adding ferrous solution in the reactor through a micro capillary (Eppendorf microloader).
For imaging with electron microscopy, particles were adsorbed from the aqueous suspensions to the carbon film Cu grids. After removal of the liquid, the grids were washed with a drop of milli-Q water. The images were acquired with a Zeiss EM 912 Omega at 120 kV.
We used X-ray diffraction for mineralogical analyses. The samples were dried on a home-designed sample holder30 and analysed. Because of the very reduced amount of samples, X-ray diffraction was measured at the µ-Spot synchrotron beamline (BESSY II, Helmholtz Zentrum Berlin) with a 100 µm beam of 15 keV (ref. 31).
Change history
30 October 2013
Minor changes were made to affiliation 1 and text citations of figures.
References
Blakemore, R. Magnetotactic bacteria. Science 190, 377–379 (1975)
Schuler, D. Genetics and cell biology of magnetosome formation in magnetotactic bacteria. FEMS Microbiol. Rev. 32, 654–672 (2008)
Komeili, A. Molecular mechanisms of compartmentalization and biomineralization in magnetotactic bacteria. FEMS Microbiol. Rev. 36, 232–255 (2012)
Bell, P. E., Mills, A. L. & Herman, J. S. Biogeochemical conditions favoring magnetite formation during anaerobic iron reduction. Appl. Environ. Microbiol. 53, 2610–2616 (1987)
Frankel, R. B. & Blakemore, R. P. Precipitation of Fe3O4 in magnetotactic bacteria. Phil. Trans. R. Soc. Lond. B 304, 567–573 (1984)
Baumgartner, J. et al. Magnetotactic bacteria form magnetite from a phosphate-rich ferric hydroxide via nanometric ferric (hydr)oxide intermediates. Proc. Natl Acad. Sci. USA 110, 14883–14888 (2013)
Fdez-Gubieda, M. L. et al. Magnetite biomineralization in Magnetospirillum gryphiswaldense: time-resolved magnetic and structural studies. ACS Nano 7, 3297–3305 (2013)
Zhang, C. et al. Two bifunctional enzymes with ferric reduction ability play complementary roles during magnetosome synthesis in Magnetospirillum gryphiswaldense MSR-1. J. Bacteriol. 195, 876–885 (2012)
Uebe, R. et al. The cation diffusion facilitator proteins MamB and MamM of Magnetospirillum gryphiswaldense have distinct and complex functions, and are involved in magnetite biomineralization and magnetosome membrane assembly. Mol. Microbiol. 82, 818–835 (2011)
Siponen, M. I., Adryanczyk, G., Ginet, N., Arnoux, P. & Pignol, D. Magnetochrome: a c-type cytochrome domain specific to magnetotatic bacteria. Biochem. Soc. Trans. 40, 1319–1323 (2012)
Lohsse, A. et al. Functional analysis of the magnetosome island in Magnetospirillum gryphiswaldense: the mamAB operon is sufficient for magnetite biomineralization. PLoS ONE 6, e25561 (2011)
Murat, D., Quinlan, A., Vali, H. & Komeili, A. Comprehensive genetic dissection of the magnetosome gene island reveals the step-wise assembly of a prokaryotic organelle. Proc. Natl Acad. Sci. USA 107, 5593–5598 (2010)
Quinlan, A., Murat, D., Vali, H. & Komeili, A. The HtrA/DegP family protease MamE is a bifunctional protein with roles in magnetosome protein localization and magnetite biomineralization. Mol. Microbiol. 80, 1075–1087 (2011)
Holm, L. & Sander, C. Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 20, 478–480 (1995)
Clausen, T., Kaiser, M., Huber, R. & Ehrmann, M. HTRA proteases: regulated proteolysis in protein quality control. Nature Rev. Mol. Cell Biol. 12, 152–162 (2011)
Krojer, T., Garrido-Franco, M., Huber, R., Ehrmann, M. & Clausen, T. Crystal structure of DegP (HtrA) reveals a new protease-chaperone machine. Nature 416, 455–459 (2002)
Lee, H. J. & Zheng, J. J. PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun. Signal. 8, 8 (2010)
Smith, L. J., Kahraman, A. & Thornton, J. M. Heme proteins–diversity in structural characteristics, function, and folding. Proteins 78, 2349–2368 (2010)
Liu, J. et al. Identification and characterization of MtoA: a decaheme c-type cytochrome of the neutrophilic Fe(ii)-oxidizing bacterium Sideroxydans lithotrophicus ES-1. Front. Microbiol 3, 37 (2012)
Baumgartner, J., Bertinetti, L., Widdrat, M., Hirt, A. M. & Faivre, D. Formation of magnetite nanoparticles at low temperature: from superparamagnetic to stable single domain particles. PLoS ONE 8, 3 (2013)
Jolivet, J. P., Chaneac, C. & Tronc, E. Iron oxide chemistry. From molecular clusters to extended solid networks. Chem. Commun. (Camb.) 5, 481–487 (2004)
Baumgartner, J. et al. Nucleation and growth of magnetite from solution. Nature Mater. 12, 310–314 (2013)
Fischer, H., Neto, M., Napolitano, H. B., Craievich, A. F. & Polikarpov, I. The molecular weight of proteins in solution can be determined from a single SAXS measurement on a relative scale. J. Appl. Cryst. 43, 101–109 (2010)
Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D 66, 133–144 (2010)
Evans, P. R. Scaling and assessment of data quality. Acta Crystallogr. D D62, 72–82 (2006)
Leslie, A. G. W. & Powell, H. R. in Evolving Methods for Macromolecular Crystallography Vol. 245 (eds Read, R. J. & Sussman, J. L. ) Ch. 4 41–51 (Springer, 2007)
Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. Automated structure solution with autoSHARP. Methods Mol. Biol. 364, 215–230 (2007)
Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S. & Dodson, E. J. Efficient anisotropic refinement of macromolecular structures using FFT. Acta Crystallogr. D 55, 247–255 (1999)
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D 68, 352–367 (2012)
Fischer, A., Schmitz, M., Aichmayer, B., Fratzl, P. & Faivre, D. Structural purity of magnetite nanoparticles in magnetotactic bacteria. J. R. Soc. Interface 8, 1011–1018 (2011)
Paris, O. et al. A new experimental station for simultaneous X-ray microbeam scanning for small- and wide-angle scattering and fluorescence at BESSY II. J. Appl. Cryst. 40, s466–s470 (2007)
Li, H., Robertson, A. D. & Jensen, J. H. Very fast empirical prediction and interpretation of protein pKa values. Proteins 61, 704–721 (2005)
Acknowledgements
This work received institutional support from the Commissariat à l’Energie Atomique et aux Energies Alternatives, the Centre National de la Recherche Scientifique, Aix-Marseille University and the Max Planck Society. We are grateful to BM-30 (ESRF, Grenoble, France) and X06SA (SLS, Villigen, Switzerland) staff for technical assistance in synchrotron data collection. We thank J. Perez (SOLEIL, GIF-sur-Yvette) for help in SAXS data collection, and A. Komeili for the gift of the wild-type and ΔmamP AMB-1 strains. We acknowledge S. Siegel and C. Li for their support at the µSpot beamline of BESSY II, Helmholtz Zentrum Berlin. We thank the AFMB laboratory (Marseille) for circular dichroism measurements. M.I.S. was supported by a grant from the Eurotalent and ToxNuc-E programs. D.F. is supported by the Max Planck Society and a Starting Grant from the ERC (256915-MB2). S.R.J. and M.C.Y.C. thank the Defense Advanced Research Projects Agency (N66001-12-1-4230) for support.
Author information
Authors and Affiliations
Contributions
M.I.S., M.W., S.R.J. and P.A. performed experiments. M.I.S., P.L. and P.A. performed structure determination. W.-J.Z. prepared genomic DNA. M.I.S., M.W., S.R.J., M.C.Y.C, D.F., P.A. and D.P. analysed the data. M.I.S., D.F., P.A. and D.P. prepared the manuscript. D.F, M.C.Y.C., P.A. and D.P. supervised the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Example of the quality of the 2mFobs − DFcalc electron density map.
Electron density maps are contoured at 1σ around the open (top) and closed (bottom) dimers. In both cases, one monomer is coloured in gold and the other in white.
Extended Data Figure 2 Sequence alignment of MamP proteins from different MTB and structural annotations discussed in the text.
Black circles, acidic residues creating a hydrogen-bond network at the bottom of the crucible; green circles, acidic residues creating a hydrogen-bond network on the side of the crucible together with the propionate moieties of the haem from the MC1 domains; H, polar residues connecting H93 side chain to the exterior of the protein. Secondary structures are indicated at the bottom of the alignment.
Extended Data Figure 3 pH-dependent oligomeric assembly of MamP.
a, Gel filtration of MamP using different buffers at different pH indicating a pH-dependent tetramer/dimer equilibrium. SAXS experiments confirm the presence of this equilibrium (see Methods). b, Circular dichroism measurement of MamP at pH 5 and 9 showing that there is no major structural rearrangement between the two pH values. c, The construction of the two different dimers of MamP (one in green, the other in cyan) starting from the two molecules in the asymmetric unit. These two molecules in the asymmetric unit are related by a non-crystallographic symmetry (NCS represented in magenta) axis. The two dimers (AC and BD) are generated using the twofold symmetry axis of the crystal (represented in black). The two dimers are therefore symmetric but they slightly differ, mainly in the orientation of two side chains of important residues located in the crucible (see Extended Data Fig. 4) supporting the notion of an ‘open’ (AC) and a ‘closed’ (BD) dimer. d, Superimposition of the two symmetric open and closed dimers. The root mean square distance between the Cα positions of 176 superimposed residues is 0.51 Å, showing that there is no major structural difference between the two states.
Extended Data Figure 4 Putative hydrogen-bond network in the crucible of the two MamP dimers and protonation states at pH 9 deduced from pKa calculations of protonable residues.
a, Putative hydrogen-bond network and protonation states of the conserved acidic residues in the crucible of the AC (open) dimer of MamP. b, Putative hydrogen-bond network and protonation states in the BD (closed) dimer of MamP. Note the small reorientation of the side chains of E193 and E123 and the repercussion on the calculated charge and, ultimately, the stabilization of two water molecules at the dimeric interface: in the open dimer, the two side chains could stabilize two water molecules (W) through two putative hydrogen bonds, which is not the case in the closed dimer. In the Fe(ii) soaking experiment, the anomalous electron density extends towards these two water molecules in the open dimer, whereas it is not visible in the closed dimer, indicating that this last conformation is not compatible with iron binding. All the putative hydrogen bonds drawn here are below 3.2 Å distance. c, Calculated pKa values of conserved residues at the bottom of the crucible in the open and closed dimers. These pKa values were calculated using PROPKA32 and the charge was deduced assuming a pH of 9.
Extended Data Figure 5 Detail of a putative hydrogen-bond network of conserved polar residues and water molecules connecting the side chain of H93 at the bottom of the crucible to the exterior of the protein.
One monomer is coloured in a ramp from blue (N-Ter) to red (C-Ter), the other one is coloured in white and rendered transparent for clarity.
Extended Data Figure 6
Size distribution of crystals determined by transmission electron microscopy. Wild type (420 particles, 28 cells), ΔmamP (425 particles, 38 cells), ΔmamP + mamP (320 particles, 29 cells), ΔmamP + mamPΔacid (528 particles, 46 cells).
Extended Data Figure 7 Western blot of MamP to determine expression of MamP and MamP mutant complements.
The lanes are loaded as follows: whole cell extract of (1) wild type AMB-1, (2) ΔmamP, (3) ΔmamP + mamP, (4) ΔmamP + mamPΔacid. The antibodies were raised to a peptide of MamP of approximately 20 amino acids from strain AMB-1 (QLEGAPMILAGPRPHGYR) in rabbits by ProSci (Poway). Western blot analysis of MamP was done for each of the three biological replicates used to collect Cmag and TEM statistics. These images are representative of those collected for all three replicates.
Extended Data Figure 8 TEM images indicating the presence of electron dense particles when MamP is present.
a, Typical TEM image of the synthesis in presence of the protein. The image shows the presence electron-dense particles, probably the magnetite found by X-ray diffraction together with poorly crystalline particulate matter. b, Typical TEM image of the synthesis in the absence of MamP. Only a gangue of iron ions, probably condensate from the solution while preparing the TEM grids, can be detected. These images are representative of those collected during the experiment.
Extended Data Figure 9 Time-resolved analysis of the mineralization synthesis followed by X-ray diffraction.
Reference peaks of ferrihydrite, magnetite and sodium chloride used as salt during the synthesis and their relative intensity are indicated.
Rights and permissions
About this article
Cite this article
Siponen, M., Legrand, P., Widdrat, M. et al. Structural insight into magnetochrome-mediated magnetite biomineralization. Nature 502, 681–684 (2013). https://doi.org/10.1038/nature12573
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature12573
This article is cited by
-
Bioinspired macromolecular templates for crystallographic orientation control of ZnO thin films through zinc hydroxide carbonate
Polymer Journal (2022)
-
Redox control of magnetosome biomineralization
Journal of Oceanology and Limnology (2021)
-
MamY is a membrane-bound protein that aligns magnetosomes and the motility axis of helical magnetotactic bacteria
Nature Microbiology (2019)
-
Magnetosomes: biogenic iron nanoparticles produced by environmental bacteria
Applied Microbiology and Biotechnology (2019)
-
Living cell synthesis of CdSe quantum dots: Manipulation based on the transformation mechanism of intracellular Se-precursors
Nano Research (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
